
Abstract
Abstract
Omics is a form of high-throughput systems science. However, taxonomies for omics studies are limited, inviting us to rethink new ways in which we classify, prioritize, and rank various omics systems science studies. In this overarching context, the genome-wide study approaches have proliferated in number and popularity over the past decade. However, their hierarchy is not well organized and the development of attendant terminology is not controlled. In the present study, we searched the literature in PubMed and the Web of Science databases published from March 1999 to September 2016 using the keywords, including genome-wide, association, whole genome, transcriptome-wide, metabolome, epigenome, and phenome. We identified the whole genome study approaches and sorted them according to the omics technology types (genomics, proteomics, and so on) and hierarchy. Thirty-four studies from over 90 publications were sorted into 10 omics groups: DNA level, transcriptomics, proteomics, interactomics, metabolomics, epigenomics, miRNomics/ncRNomics, phenomics, environmental omics, and pharmacogenomics. We suggest here modifications of terminology for study approaches, which share the same acronyms such as EWAS for epigenome-wide association and environment-wide association studies, and MWAS for methylome-wide association and metabolome-wide association studies. Taken together, our study presented here provides the first systematic review and analyses of whole genome approaches and presents a baseline for further controlled terminology development, with a view to a new taxonomy for omics and multi-omics studies in the future. Finally, we call for greater dialogue and collaboration across diverse omics knowledge domains and applications, for example, across plants, animals, clinical medicine, and ecology.
Introduction
O
In this overarching context, the genome-wide (whole genome) study approaches have proliferated in number and popularity over the past decade. One of the first developed whole genome study approaches was genome-wide association study (GWAS), which tries to find significant associations between single-nucleotide polymorphisms (SNPs) and studied traits (Manolio, 2010). Despite promising at the beginning, several reported findings of GWAS have only small effects on observed phenotypes and in addition explain only a small proportion of the variance in a trait that is known to be influenced by genetics. Therefore, several complimentary approaches for GWAS have been developed such as genome-wide association interaction study (GWAI) (Gusareva et al., 2014), pathway-based GWAS (Jiao et al., 2015), metabolome-wide association study (MWAS) (Chadeau-Hyam et al., 2010), epigenome-wide association study (EWAS) (Flanagan, 2015), and phenome-wide association study (PheWAS) (Hebbring, 2014). The development of numerous whole genome study approaches has been enabled by the development of several experimental as well as computational technologies.
For example, the development of next-generation sequencing (NGS) enables to sequence DNA and RNA much more quickly and cheaply and thus has laid the foundation for the development of several study approaches (Metzker, 2010). Furthermore, many modifications of NGS such as bisulfite sequencing (Li and Tollefsbol, 2011) and chromatin immunoprecipitation sequencing (ChIP-seq) (Hoffman and Jones, 2009) have been developed. However, hierarchy of whole genome study approaches and technologies is currently not organized, and thereby, the development of terminology is also lagging behind. As a result, certain study approaches have been named with different terms and in addition the same acronyms are used for different approaches. Therefore, problems in literature searching are occurring, and novel developed study approaches do not have any guidance in terminology development, which leads in even greater disorganization.
The aim of the present study was, therefore, to provide an overview of the genome-wide study approaches and establish a baseline for terminology development. We have organized study approaches hierarchically and attempted to organize the current terminology.
Materials and Methods
We searched systematically the types of genome-wide study approaches from scientific literature in PubMed (www.ncbi.nlm.nih.gov/pubmed) and Web of Science (http://apps.webofknowledge.com) from March 1999 to September 2016. The articles have been identified using the keywords such as genome-wide, association study, whole genome, whole transcriptome, transcriptome-wide, metabolome, epigenome, phenome, interactions, and environment. Collected study approaches have been sorted according to the omics types in 10 groups: genomics/DNA level, transcriptomics/RNA level, proteomics, interactomics, metabolomics, epigenomics, miRNomics/ncRNomics, phenomics, environmental omics, and pharmacogenomics. In addition, we have extracted the names of study approaches and organized them hierarchically. Furthermore, we have briefly described collected study approaches.
Results
We have collected over 90 publications describing study approaches and methods on a genome-wide scale. Study approaches were then sorted by omics type (Fig. 1). Examples of the reports using whole genome study approaches found in the literature, their acronyms, and references are presented in Table 1. The most common experimental techniques that are used at different omics levels are listed in Table 2. Based on the collected studies, we created hierarchy organization of genome-wide study approaches, which is presented in Table 3. This suggested list of study approaches now serves as a baseline for extending and complementing the list using approaches in future reports.

Presentation of different study approaches and omics types used in genomics. Studies could be performed on a single locus or on a genome-wide scale.
miRNA, microRNA; QTL, quantitative trait locus; SNP, single-nucleotide polymorphisms.
Analysis of the collected literature revealed a large disorganization in terminology in this research area. For example, certain study approaches have been named with different names, some study approaches share the same acronym, and some of them do not have an acronym. Therefore, to arrange this disorganization, we have made some suggestions for changes of terminology.
Study approaches used for whole genome analyses
The term whole genome analysis is used in two cases; WGA in a narrow sense focuses on a genome at the DNA level, on the contrary, in a broad sense, WGA considers all omics approaches and a genome as a whole.
DNA level
Whole genome sequencing (genome-wide sequencing)
Whole genome sequencing (WGS) is a process of determining the entire DNA sequence of an organism's genome. The result is the precise order of nucleotides within DNA molecules of the entire genome. Many different methods of WGS exist. The first developed method was traditional WGS that includes cloning of DNA fragments in microbial cells and sequencing of these fragments by using Sanger dideoxy sequencing technique. Newly developed technologies replaced the traditional methods for WGS. These methods are known as next-generation WGS and are cell-free methods.
The cloned fragments are sequenced by using NGS methods. Most of the WGS methods have the same basic steps: (1) breaking of the DNA molecules of a genome up into thousands to millions of more or less random, overlapping small segments, (2) sequencing of each small segment, (3) computationally analyzing and finding of the overlap among the small segments where their sequences are identical; reassembling of sequenced fragments into contigs, and (4) overlapping of ever larger contigs until all the sequence is completed (Bick and Dimmock, 2011; El-Metwally et al., 2013; Pareek et al., 2011).
There are two main strategies of WGS, including de novo genome sequencing and whole genome resequencing. De novo sequencing is initial determination of the primary genomic sequence of a specific organism and it is crucial for detailed genetic analysis of any organism. On the contrary, whole genome resequencing is one of the most frequently used applications of NGS technology and it is used for the identification of SNPs, indels, copy number variations, and structural variations, as well as for identification of multiple individuals or strains (Zhou et al., 2010). The bioinformatics analysis that follows the process of DNA resequencing consists of three basic steps: alignment to reference genome, identification of sequence variations (variant calling), and filtering and annotation of identified variants (Dolled-Filhart et al., 2013).
The genome sequencing projects were long limited only to model organisms, but the progress in high-throughput sequencing technology and the development of many bioinformatics tools have expanded the field of use (Ekblom and Wolf, 2014). Many successful applications of WGS in identification of the etiology of complex diseases have been reported. For example, the results of WGS have helped in choosing the right therapy approach for neoplastic and nonneoplastic diseases and in various aspects of reproductive health (Chrystoja and Diamandis, 2014).
Whole exome sequencing (exome-wide sequencing)
Whole exome sequencing (WES) is used for sequencing of coding regions or the exons of known genes, which constitute about 1–2% of the human genome (Volk et al., 2015). WES covers up more than 95% of the exons, which contain 85% of disease-causing mutations. This is one of the reasons that sequencing of entire human exome has the potential to discover the main genetic causes for rare mostly monogenic diseases (Mendelian disorders) as well as disease predisposing variants in diseases. WES has been successfully used to identify the causative variants in several conditions, including hearing loss, intellectual disabilities, autism spectrum disorders, retinitis pigmentosa, diabetes, cardiovascular disease, hypertension, obesity, and cancer. Furthermore, in comparison to WGS, it is also less costly and consequently more common in clinical and diagnostic tests (Rabbani et al., 2014).
GWAS (whole genome association study)
GWAS is a study approach that involves examination of many common genetic variants, usually SNPs, in different individuals across the genomes to find genetic variants associated with a particular phenotypic trait or disease (Manolio, 2010). Studies using GWAS approach have identified many genetic variants associated with human diseases such as age-related macular degeneration, Crohn's disease, schizophrenia, multiple sclerosis, ankylosing spondylitis, and rheumatoid arthritis (Hebbring, 2014; Manolio, 2010).
There are two main types of GWAS design: (1) the case/control studies that compare the DNA of individuals with/without the disease and (2) the quantitative trait association studies (Dubé and Hegele, 2013). Most commonly used genotyping technology is chip-based microarray technology, which can genotype over one million SNPs across the human genome in a single assay, followed by statistical analysis consisting of independent single-locus statistical tests that examine each SNP for association to the trait (Bush and Moore, 2012). Results of GWAS are usually represented with Manhattan plot (Dubé and Hegele, 2013).
Even though GWASs have identified thousands of genes and genetic variants that contribute to complex traits, especially diseases, there are still a lot of limitations to overcome. For instance, difficulties in obtaining statistical significant results, GWAS's results often just partly explain the heritable variation, and the vast majority of GWAS-significant SNPs are intergenic, which means identifying and characterizing of these SNPs is even more challenging (Hebbring, 2014).
Whole genome comparative quantitative trait locus analysis
Whole genome comparative quantitative trait locus (QTL) analysis is a study approach that compares the locations of QTL controlling studied traits in at least two species. For example, a whole genome comparative QTL analysis has been performed for fruit weight, pericarp thickness, and fruit shape in tomato and pepper. They have identified 95 QTL, including two QTL for fruit weight that was common in both species (Chaim et al., 2006).
RNA level, transcriptomics
Whole transcriptome analysis, transcriptome-wide analysis, genome-wide transcriptome analysis
Whole transcriptome analysis (WTA), also known as transcriptome-wide analysis or gene expression profiling, is a study approach that measures the expression of thousands of genes at once. The aim of WTA is to capture coding and noncoding RNA (ncRNA) to quantify gene expression and identify heterogeneity of gene expression profiles in cells, tissues, organs, and even a whole body. It is used for decoding genome structure, characterizing and annotating genes/genomes previously revealed by DNA sequencing, identifying genetic networks of cellular, physiological, biochemical, and biological systems, and determining molecular biomarkers that respond to diseases, pathogens, and environmental challenges (Jiang et al., 2015). The most common techniques that are used for gene expression profiling are DNA microarrays and RNA sequencing (RNA-seq) (Mantione et al., 2014; Qian et al., 2014).
Whole transcriptome sequencing, whole transcriptome shotgun sequencing, RNA-seq
Whole transcriptome sequencing (WTS), also known as whole transcriptome shotgun sequencing (WTSS) or RNA-seq, is an application of any type of NGS techniques to study RNA (Chu and Corey, 2012). This study approach has become more available with the development of NGS techniques. RNA-seq technology is used for (1) the quantitative analysis of transcript expression levels, (2) the annotation of transcripts, (3) the detection of low abundance transcripts, (4) the study of mRNA processing, (5) the analysis of alternative splicing, and (6) the study of polyadenylation (Marguerat and Bähler, 2010). Sequencing process is almost the same for RNA-seq and DNA sequencing, but the library preparation and the data analysis are different. The preparation of RNA-seq library usually includes reverse transcription and labeling with adapter sequences (Chu and Corey, 2012; Mantione et al., 2014).
Proteomics
Whole proteome analysis
Proteomics is the large-scale study of proteins and their characteristics, including expression level, post-translational modifications, and interactions. The aim of this study approach is to obtain a global integrated view of disease processes, cellular processes, and networks at the protein level (Blackstock and Weir, 1999). There are several methods for the detection, identification, and functional investigation of proteome such as two-dimensional gel electrophoresis (Rabilloud et al., 2010), mass spectrometry (Lesur and Domon, 2015), and protein microarrays (Uzoma and Zhu, 2013). Protein microarrays, known also as proteome microarrays, are offering a wide variety of applications and have been used to study several types of interactions, such as protein–protein, protein–DNA, protein–lipid, protein–drug, protein–receptor, and antigen–antibody interactions (Hall et al., 2007), as well as for identification of protein posttranslational modifications such as phosphorylation, ubiquitylation, acetylation, and S-nitrosylation (Zhu and Qian, 2012). For example, the human proteome microarray has been used to identify proteins that interact with alpha A-crystallin (Fan et al., 2014). Furthermore, human S-nitrosoproteome has been analyzed using high-density protein microarray chip resulting identification of 834 potentially S-nitrosylated human proteins (Lee et al., 2014).
Interactomics
Interactomics include several study approaches that consider various types of interactions such as DNA–DNA, DNA–protein, RNA–protein, and protein–protein.
DNA–DNA interactions
Published reports describe two types of DNA–DNA interactions, physical interactions and statistical (epistatic) interactions.
Physical interactions
Genome-wide analysis of physical interactions between DNA molecules
Chromosome conformation capture (3C) is a method that detects short- and long-range physical DNA–DNA interactions. It was first developed for analysis of chromosomes conformation in yeast (Dekker et al., 2002). This technique has been rapidly adapted as a standard research tool for studying chromosomal interactions (Li, 2016). An extension of 3C method is Hi-C method, which is able to identify long range interactions in an unbiased genome-wide manner. It is a method used for studying the three-dimensional architecture of genomes. The three-dimensional organization of a genome plays important roles in regulation of genes and the functional state of the cell, but yet it is poorly understood how genomes are spatially organized, and how this affects gene expression. The Hi-C method provides a promising tool for deciphering the relationship between chromosome organization and genome activity and it will contribute to the understanding of genomic processes such as transcription and replication (Lieberman-Aiden et al., 2009; van Berkum et al., 2010).
Statistics/epistatic interactions
Several of reported discoveries from GWAS have small effects on observed phenotype and explain only a small proportion of the variance in a trait that is known to be influenced by genetics (Hibar et al., 2015). It has been hypothesized that multivariate approaches such as analyses of genetic interactions may resolve the problem of “missing heritability” observed in GWAS analysis. Therefore, several study approaches have been reported to identify DNA–DNA interactions on a genome-wide scale, including genome-wide interaction analysis (GWIA) (Hibar et al., 2015), GWAI (Gusareva et al., 2014), genome-wide search for SNP–SNP interactions (Murk and DeWan, 2016), and genome-wide gene–gene interaction analysis (Chu et al., 2014).
Methods developed for detection of interactions are typically focused on SNP interactions (epistatic interactions) (Goudey et al., 2013). These methods have been used for various analyses, for example: testing all possible SNP–SNP interactions affecting regional brain volumes (Hibar et al., 2015), identification of Alzheimer's disease associated interacting SNP pairs (Gusareva et al., 2014), identification of SNP–SNP interactions associated with 10 common human diseases (Murk and DeWan, 2016), and identification of epistatic gene pair that was significantly associated with the risk of developing lung cancer (Chu et al., 2014). Although improved processor speeds have lead two-way interactions analyses in common use, there are still limitations due to underpowered statistical approaches (Goudey et al., 2013; Hibar et al., 2015). Currently, the software tools development is heading toward developing algorithms, which will also allow analyzing three- or more-way interactions (Goudey et al., 2013).
DNA–protein interactions
Genome-wide analysis of protein–DNA interactions
Genome-wide analysis of protein–DNA interactions is a study approach that identifies interactions between proteins and specific regions of the genome. ChIP is a common method for detecting interactions between a protein and DNA sequence in vivo. In addition, this method has been combined with DNA microarrays (ChIP–chip) and NGS (ChIP-seq) and thus has enabled genome-wide identification of DNA-binding sites for numerous nuclear proteins (Hoffman and Jones, 2009; Kim and Ren, 2006; Pugh and Gilmour, 2001).
Genome-wide mapping of transcription factor binding sites
For understanding of gene expression regulation, it is also crucial to identify gene-specific transcription factors and transcription factor binding sites (TFBS). Genome-wide mapping of TFBSs is a study approach that identifies specific genomic regions for TFBSs. Genome-wide maps of TFBSs in primary tissues can increase our knowledge about genome function, transcriptional regulation, and genetic alterations that contribute to disease risk (Savic et al., 2013). For example Hif-1α binding sites in the zebrafish genome have been identified using ChIP-seq method, enabling development of zebrafish as an emergent model organism for research into the hypoxic response in a physiological context (Greenald et al., 2015).
RNA–protein interactions
Genome-wide analysis of RNA–protein interactions
Genome-wide analysis of RNA–protein interactions is a study approach that identifies the interactions between RNA binding proteins (RBPs) and specific RNA molecules (Barkan, 2009). Common methods for studying interactions between proteins and RNA molecules are crosslinking and immunoprecipitation (CLIP) (Ule et al., 2003), as well as its variations, including photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP) (Hafner et al., 2010a), and individual-nucleotide resolution UV CLIP (iCLIP) (König et al., 2010). For instance, transcriptome-wide identification of RBP and microRNA (miRNA) target sites has been performed using PAR-CLIP method (Hafner et al., 2010b).
Protein–protein interactions
Genome-wide protein–protein interaction analysis
Genome-wide protein–protein interaction (PPI) analysis is a study approach that combines data from different sources to predict PPIs and establish PPI networks (Lv et al., 2015). PPIs are generally identified by pull-down experiments or other similar techniques. However, this approach is too slow as well as too expensive to meet the goal of identifying all the PPIs necessary to understand the functional and dynamic properties of the cell. Therefore, several computational methods have been developed to compliment experimental methods. They can efficiently integrate data from several sources to make predictions of the probability of interaction between two proteins (McDowall et al., 2009). The main aim of genome-wide PPI analysis is to investigate functions of proteins and thus provide better understanding of the biological mechanism of proteins, protein complexes, and their molecular interactions (Lv et al., 2015).
Network-based studies
Network-based analysis of GWASs
Network-based approaches have become powerful tools for studying complex diseases. It is known that genes, gene products, and small molecules interact with each other and form a complex interaction network (Cho et al., 2012). The aim of network-based analysis of GWASs is the understanding of the way multiple modestly-associated genes interact to influence a phenotype (Chang et al., 2015a). Different intermolecular interactions exist such as PPIs, protein–DNA interactions, and RNA interactions. These interactions are represented as networks with nodes that symbolize molecules and links that symbolize their interactions.
There are two types of interaction networks: physical and functional. Physical networks represent contacts between proteins, on the contrary, functional interactions networks aim to connect genes with similar or related functions. In studies of complex diseases, researchers are usually focused on groups of related genes, referred to as modules or subnetworks. Often it is easier to predict the function of a module than the function of a single gene. Modules are identified by using diverse genomics information, including genotypic data and expression profiles identified in disease samples (Cho et al., 2012). Several software tools have been developed to generate networks and modules, including network interface miner for multigenic interactions (NIMMI) (Akula et al., 2011) and ARACNE (Margolin et al., 2006). Most network-based analyses combine GWAS data with PPI data (Akula et al., 2011; Baranzini et al., 2009; Consortium, 2013; Zhang et al., 2015a). Simplified workflow of network-based study approach is presented in Figure 3a.
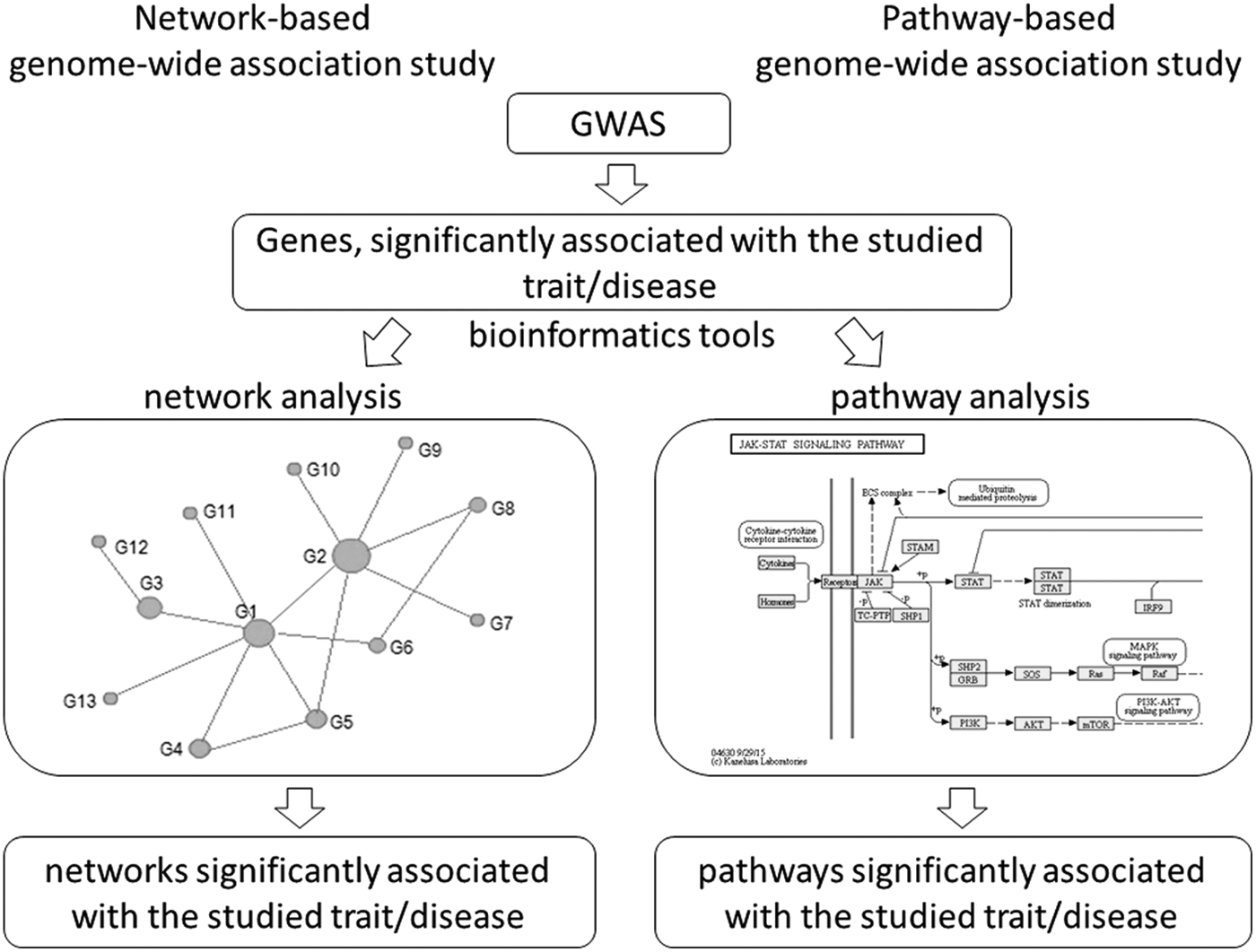
The comparison of the network-based and pathway-based genome-wide association study approaches.
Protein interaction network-based analysis
Protein interaction network-based analysis (PINBA) of GWAS data is a network-based method for identifying susceptibility genes, which investigates whether a set of genes with related function is jointly associated with a trait or disease of interest. PINBA combines GWAS data and prior biological knowledge about protein interactions to determine associability (Yu et al., 2014). Protein interaction network-based pathway analysis (PINBPA) has been used to investigate missing heritability in multiple sclerosis. They have combined two largely independent GWAS multiple sclerosis data sets and discovered that proteins encoded by genes that contain risk variants are more likely to interact with each other and take part in the same or related pathways (Consortium, 2013).
Integrative network-based association study
Integrative network-based association study (INAS) integrates different types of data, including GWAS data, protein–protein, and protein–DNA interactions, to generate regulatory networks and thus provides understanding of the roles of genetics and epigenetics in disease predisposition and etiology (Califano et al., 2012). For example, Lage et al. have performed systematic analysis of human protein complexes consisting of gene products involved in several categories of human diseases to create a phenome–interactome network. They have integrated quality-controlled data of human protein interactions and validated a computationally derived phenotype similarity score (Lage et al., 2007).
Pathway-based studies, gene set enrichment analysis
Pathway-based GWAS, pathway-based analysis
Pathway-based study approaches examine whether a group of related genes in the same functional pathway are together associated with a studied trait (Wang et al., 2010). Several different names have been used for this study approach such as pathway-based GWAS (Wang et al., 2010), pathway-based analysis (PBA) (Chang et al., 2015b), and pathway-wide association study (PWAS) (Califano et al., 2012). The term “pathway” in GWASs usually means a gene set, rather than an interconnected biological process (Jiao et al., 2015). Pathway-based studies are complimentary approaches for GWAS. GWASs have focused on the analysis of single markers and thus have often overlooked genetic variants with relatively small effect sizes.
On the contrary, pathway-based GWAS can detect the additive effects of multiple minor genes. Therefore, it has a better chance of identifying novel genes or sets of genes and mechanisms that are involved in disease pathogenesis. Pathway-based GWASs usually examine a collection of predefined gene sets for pathways based on prior biological knowledge. In recent years, a variety of methods have been developed for pathway-based association analysis. The disadvantage of PBA is that this approach sees pathway as a single unit and thus cannot detect a small portion of the pathway or other new combinations of genes that may be associated with a trait (Chang et al., 2015b). Simplified workflow of pathway-based study approach is presented in Figure 3b.
Metabolomics
Metabolite-based GWAS (metabolome-based GWAS)
Metabolite-based GWAS (mGWAS) (metabolome-based GWAS) is a combination of GWAS and metabolomics (Luo, 2015). It is defined as a genome-wide association study with metabolic traits as phenotypic traits (Gieger et al., 2008). The metabolite spectrum and the metabolites' quantities may be viewed as the metabolic phenotype or metabotype. If the metabotype provides a link between gene sequence and visible phenotypes, metabolites can be used as biomarkers for prediction of the traits (Wen et al., 2014). The first mGWAS has investigated associations between genetic variants and metabolite profiles in human serum (Gieger et al., 2008). Using this approach in plants, they have discovered which DNA variants have significant impact on metabolic changes. This has extended the knowledge on the genetic contribution to plant metabolism and on the by now undiscovered biochemical pathways or pathway interactions (Luo, 2015).
Metabolome-wide association studies
MWAS is a study approach that investigates associations between metabolic phenotypes and disease risk and thus can be used for identification of disease-related biomarkers (Chadeau-Hyam et al., 2010). MWAS has some similarities with GWAS, such as case/control approach of the study, high throughput, discovery of novel associations, large complex data arrays, and potential for false-positive association (Bictash et al., 2010; Osborn et al., 2013). It starts with metabolic profiling, which applies metabolomics techniques such as nuclear magnetic resonance spectroscopy and mass spectrometry, liquid chromatography–mass spectrometry, gas chromatography–mass spectrometry, flow injection analysis mass spectrometry to measure hundreds or thousands of metabolites in cells, biofluids, or tissues (Chadeau-Hyam et al., 2010; Petersen et al., 2012). The next step is statistical analysis of the data, which detects statistically significant relationships between molecular variables and phenotype (Chadeau-Hyam et al., 2010).
Epigenomics
Epigenome-wide association study
EWAS is a study approach that is used to analyze epigenetic marks, which are often DNA methylation and histone modifications (Flanagan, 2015). Using EWAS approach, numerous significant associations have been identified for several diseases, especially in the context of the cancer, exposures, and lifestyle factors (Flanagan, 2015; Rakyan et al., 2011).
Methylome-wide association study
MWAS is one of the subclasses of EWAS. DNA methylation is associated with gene silencing (Bird, 2002). Methylation studies are a promising compliment to genetic studies. First, the methylation affects gene expression, and therefore, the knowledge about DNA methylation can contribute to understanding of disease etiology. It has been already discovered that dysregulation of DNA methylation has been associated with many human diseases such as Alzheimer's and Parkinson's diseases, systemic lupus erythematosus, cardiovascular disease, and several types of cancer. Second, methylation is also affected by many factors that can affect the development of diseases such as age, sex differences, and genotype environment interactions. Finally, methylation sites are potential new drug targets (Aberg et al., 2012; Chen et al., 2013). The most common techniques for profiling DNA methylation are sequencing and array-based profiling technologies (Rakyan et al., 2011).
Whole genome bisulfite sequencing (genome-wide bisulfite sequencing)
Bisulfite genomic sequencing is technology for detection of DNA methylation and provides a qualitative, quantitative, and efficient approach to identify 5-methylcytosine at single base-pair resolution (Li and Tollefsbol, 2011). Whole genome bisulfite sequencing (WGBS or WGBS-seq) is a combination of bisulfite conversion of genomic DNA and NGS (Lee et al., 2013). Unmethylated cytosines are after the treatment of sodium bisulfite converted into uracils, while methylated cytosines are immune to this conversion and remain as cytosines (Li and Tollefsbol, 2011). Next step is sequencing that usually includes the pyrosequencing method, which is a quantitative method for DNA methylation analysis (Lee et al., 2013).
WGBS-seq has been successfully used to map the complete methylomes of several human embryonic stem cell (ESC) lines, human peripheral mononuclear cells, and hematopoietic progenitor cells. WGBS-seq analyses have also contributed to discovery of large partially methylated domains in cancer cells. Although there are many advantages of WGBS-seq, this approach is not economically feasible for the large sample sizes. Therefore, other alternative sequencing approaches developed such as targeted bisulfite sequencing (Lee et al., 2013), methyl-CpG binding domain (MBD) protein-enriched genome sequencing (MBD-seq), and methylated DNA immunoprecipitation (Chen et al., 2013; McClay et al., 2014).
Genome-wide analysis of histone modifications
Histone modifications are post-translation modifications of histone proteins, including methylation, phosphorylation, acetylation, ubiquitylation, and SUMOylation. These modifications can impact gene expression by directly altering chromatin structure or on the other hand by providing binding sites for the recruitment of other nonhistone proteins to chromatin (Zhang et al., 2015b).
Genome-wide analysis of histone methylation
Methylation of histone N-terminal tails can act to regulate chromatin states. Methylation of lysines 4, 36, and 79 of histone H3 is associated with euchromatin, while the methylation of histone H3 lysines 9, 27, and histone H4 lysine 20 is associated with heterochromatin and gene silencing (Barski et al., 2007; Guenther et al., 2007).
Genome-wide analysis of histone methylation on two histone H3 lysine residues (H3K4me3 and H3K27me3) and gene expression profiles in naive and memory CD8+ T cells has been performed, and the existence of correlation between gene expression and the amounts of H3K4me3 (positive correlation) and H3K27me3 (negative correlation) across the gene body has been identified. The analysis of histone methylation has been performed using ChIP-seq and the global gene expression profile by the human whole genome chip. The correlation between the amount of H3K4me3 or H3K27me3 with mRNA has been confirmed by real-time polymerase chain reaction (PCR) or quantitative PCR (qPCR) and reverse transcription PCR (RT-PCR) (Araki et al., 2009).
Genome-wide analysis of histone acetylation
Epigenetic alternations, including histone acetylation and deacetylation, are important parts of gene regulation. Acetylation of histone H3 lysines 9 and 14 is associated with euchromatin and active gene transcription (Araki et al., 2009). Genome-wide histone H3 acetylation patterns in acute myeloid leukemia (AML) have been analyzed using ChIP-chip in a group of patients with AML and controls to identify epigenetic alterations in AML. They have discovered that in AML, core promotor regions have lower histone H3 acetylation levels compared with CD34+ progenitor cells (Agrawal-Singh et al., 2012). In addition, Liu et al. (2015) have performed genome-wide analysis of histone acetylation dynamics during mouse ESC (mESC) neural differentiation by ChIP-seq and revealed that during mESC neural differentiation the histone H3 acetylation level is increased on the neural gene loci while decreased on the neural inhibitory gene loci.
Genome-wide nucleosome mapping
The availability of genome-wide nucleosome maps has contributed to knowledge about the factors that affect nucleosomes' position and how this consequentially influences and regulates various cellular processes, including regulation of gene expression, chromosome segregation, DNA replication and repair, and recombination. These novel findings represent a whole picture of complicated interactions between DNA sequence, various regulatory processes, and the three-dimensional chromatin structure. Most of the nucleosome mapping techniques share the same basic principles. First, the unprotected linker DNA is hydrolyzed by using micrococcal nuclease (MNase), followed by isolation, purification, and analysis of undigested, nucleosomal DNA fragments. Several techniques provide different levels of resolution and accuracy for nucleosome positioning analysis. Genome-wide studies often use microarray hybridization or high-throughput sequencing to identify nucleosome positions. Nucleosomes have been mapped both in vivo and in vitro. In vivo studies represent the chromatin organization in living cells; on the other hand, in vitro studies focus only on DNA-encoded sequence preferences and exclude the effects of other trans factors (Jansen and Verstrepen, 2011).
miRNomics, ncRNomics
ncRNA are functional RNAs that are not transcribed into proteins. According to the 200 bp limit, they are divided to short and long ncRNA (lncRNAs). miRNA are around 22 bp in length. They bind to different target regions and silence or activate expression of target genes (Kunej et al., 2012). They play an important role in regulation of several processes, including organismal development and establishment and maintenance of tissue differentiation (Pritchard et al., 2012). lncRNAs are still poorly characterized, but recently it has been shown that they play an important role in several biological processes, including gene expression, immune response, cellular development, and metabolism through comprehensive mechanisms (Yarmishyn and Kurochkin, 2015; Zhu et al., 2014).
Genome-wide miRNA profiling (whole genome miRNA profiling)
Genome-wide miRNA profiling is a study approach that measures the expression of miRNA molecules in selected cells or tissues. The importance of miRNA in gene expression regulation has led in development of miRNAs as biomarkers for variable molecular diagnostic applications, in cancer, cardiovascular and autoimmune diseases, and also in development of miRNA-based methods for forensic analysis (Pritchard et al., 2012). The most common approaches that are used for genome-wide miRNA profiling are miRNA microarrays and NGS (RNA-seq) (Liu et al., 2004; Munker and Calin, 2011; Pritchard et al., 2012). RNA-seq has several advantages over other approaches, including discovery of novel miRNAs and identification of low-abundance miRNAs (Morin et al., 2008).
Genome-wide analysis of lncRNA expression
Genome-wide analysis of lncRNA expression is a study approach that measures the expression of lncRNA molecules in selected cells or tissues. Most common methods that are used for genome-wide analysis of lncRNA expression are microarray assay, usually validated by qPCR analysis and high-throughput sequencing technologies (Sui et al., 2013; Yarmishyn and Kurochkin, 2015).
Phenomics
Phenome-wide association study
PheWAS is a study approach that investigates the association between SNPs and a wide spectrum of phenotypes, known as phenome (Pendergrass et al., 2012). It is a complementary approach to GWAS, known also as “reverse GWAS” (Denny et al., 2013; Pendergrass et al., 2012). GWAS is a phenotype-to-genotype approach, which means that it begins with a specific phenotype to test for associations over a wide spectrum of genetic variants across the genome; on the other hand, PheWAS uses the reverse approach—genotype-to-phenotype strategy, which means that it starts with a genotype to test for associations over a wide spectrum of human phenotypes. The comparison between PheWAS and GWAS approaches is presented in Figure 3.
Most PheWAS have in common the use of an electronic medical record (EMR) to define the phenome. EMR includes a number of data such as prescription records, family histories, laboratory and imaging test results, physician notes, procedure codes, and most importantly the International Classification of Disease (ICD) codes, which are internationally standardized codes used for defining the disease status. The patients coded for a specific ICD9 code are usually identified as “cases” for that respective code. On the contrary, the patients not coded for a specific ICD9 code become “controls.” The successfulness of PheWAS depends on how well the phenome can be defined. When a phenome is constructed, an association testing can begin (Hebbring, 2014). PheWASs results may be useful in detecting novel relationships between SNPs, phenotypes, as well as for the identification of pleiotropy (Pendergrass et al., 2012).

A schematic diagram of the comparison between PheWAS and GWAS approaches. GWAS, genome-wide association study; PheWAS, phenome-wide association study.
Environmental omics
Environmental omics is the application of different omics technologies to better understand the effect of environmental and genetic factors on phenotypes (Ge et al., 2013; Morrison et al., 2006).
Environment-wide association study
It has become clear that for better understanding of complex diseases the measurement of both genetic and environmental factors is crucial. Environmental-wide association study (EWAS) is a study approach that searches for environmental factors associated with disease development on a broad scale. Two EWAS for type 2 diabetes have been performed to date. Patel et al. have conducted the study by using cross-sectional epidemiological data (the National Health and Nutrition Examination Survey; NHANES). This survey collects data by querying participants about their health status, and an extensive set of clinical and laboratory tests are performed on a subset of these individuals. Several environmental characteristic are assayed, such as chemical toxicants, pollutants, allergens, bacterial/viral organisms, and nutrients.
On the contrary, Hall et al. have used four tools for measuring environmental exposures and outcome traits: the PhenX Toolkit, the Diet History Questionnaire (DHQ), the Measurement of a Person's Habitual Physical Activity, and electronic health records. The results of statistical analysis are usually presented using the Manhattan plot, with X-axis representing different environmental classes and Y-axis indicating −log10 (p-value) of the adjusted logistic regression coefficient for each of the environmental factors. Integration of EWAS with GWASs and PheWASs will increase knowledge about the complex interplay of gene and environment in complex traits and it will also elucidate the ways in which exposures modulate pleiotropy (Hall et al., 2014; Patel et al., 2010).
Genome-wide environmental interaction study, genome-wide by environment interaction study
Genome-wide environmental interaction study (GWEI) (Aschard et al., 2012) or genome-wide by environment interaction study (GWEIS) (Dunn et al., 2016) is a study approach that investigates the gene–environment (G-E) interactions on a genome-wide scale.
It is believed that complex diseases are the result of a combination of effects of genes, environment factors, and their interactions; therefore, the study of G-E interactions plays an important role in discovering disease-causing mechanisms (Aschard et al., 2012; Winham and Biernacka, 2013). It is generally considered that G-E interaction occurs if the genotype at a locus modifies the effect of an environmental factor on the phenotype, or vice versa if an environmental factor modifies the effect of a genetic variant on the phenotype. Genome-wide environmental interaction studies are conducted by performing a test of interaction with each genotyped SNP and the environmental factor. This approach has been recognized as severely underpowered for analysis of G-E interactions, and therefore, several screening approaches have been developed to reduce the number of G-E effects tested and improve power of G-E studies, including two-step/filtering methods. Genome-wide environmental interaction studies have still a lot of limitations, therefore, further development of novel approaches and software will be critical to the identification of genome-wide G-E interaction effects in complex traits (Winham and Biernacka, 2013).
Pharmacogenomics
Genome-wide mapping of drug–DNA interactions
Genome-wide mapping of drug–DNA interactions is a study approach that identifies binding sites for small molecules across the genome. In recent years, there has been great interest in the development of molecules that can selectively bind to the genome and modulate gene regulatory networks and thus increase therapeutic index and reduce off-target effects. Recently, several methods to study interactions between small molecules and nucleic acids have been developed such as chemical affinity capture and massively-parallel DNA sequencing (Chem-seq) and crosslinking of small molecules to isolate chromatin (COSMIC). COSMIC is a method that identifies polyamide binding sites across the genome. Polyamides composed of N-methylpyrrole and N-methylimidazole are molecules that can target DNA and have showed antiviral and anticancer properties (Erwin et al., 2016).
Discussion
We performed an analysis of research publications in genomics and collected existing genome-wide study approaches and organized them hierarchically. This study provides first preliminary review of the omics study approaches and presents the start of further more comprehensive analysis. The analysis of the literature revealed disorganization in terminology of genome-wide study approaches, which causes problems in literature searching and experimental work. The main challenge is that different study approaches share the same acronyms and that one study approach is named with several terms. In the future, experts from the field will need to define the differences between similar study approaches and standardize terminology. An example of the study approach that needs standardization is analysis of DNA–DNA interactions as well as network- and pathway-based studies.
In addition, we have found the following examples for study appraches that share the same acronyms. First, the acronym MWAS is used for methylome-wide association study as well as for metabolome-wide association study. Second, the acronym EWAS is used for epigenome-wide association study and environment-wide association study. Instead of changing the acronyms with a new one, extending the existing one might present a compromise. To solve this issue, we proposed some solutions by introducing new acronyms presented in Table 4.
Furthermore, we suggest that the term epigenome-wide association study (EWAS) is used as a hypernym for all types of studies of epigenetics factors in genome-wide level and that the term methylome-wide association study (MWAS) is used for studies of DNA methylation.
In addition, some study approaches can be classified in more than one omics type. For example, study approaches that analyze expression of miRNA genes may fall under miRNomics as well as transcriptomics. Furthermore, studies that investigate PPIs may be part of proteomics and interactomics.
Plausible explanation for the differences in terminology or acronym of some related approaches is that the study approaches as well as omics approaches may depend on the field of study (e.g., biomedical, microbiology, environment, food science, and cell engineering). Therefore, the scientists engaged in one particular field of study do not pay attention to study approaches in other research areas and thus lead to inconsistencies in terminology.
Conclusions and Outlook
Taken together, our study presented here provides the first systematic review and analyses of whole genome approaches and presents a baseline for further controlled terminology development, with a view to a new taxonomy for omics and multiomics studies in the future.
We have curated and organized the hierarchy of several reported genome-wide study approaches and, moving forward, suggest solutions of terminology for the same acronyms used for different methods. Moreover, our study provides an overview of the field of genomics and can be instrumental for all who are engaged in building a robust taxonomy in this research area. It provides guidelines in terminology, and, by taking our suggestions into account, the scholars in omics can develop terminology for novel study approaches in the future.
Due to rapid development and the extensiveness of the field of genomics, we did not manage to include all reported study approaches and omics types, for example, study approaches in the field of functional genomics and RNA-editomics. Furthermore, environmental omics includes several different omics approaches such as nutrigenomics and metagenomics. Therefore, the presented hierarchy organization and terminology should be further extended and complimented in the future by experts of the fields. In the present study, we were mainly focused on the different study approaches, but for even more complete overview of the field of genomics, it will be also necessary to review the methods and techniques that are used to analyze specific study approaches in more detail.
Footnotes
Acknowledgments
This work was supported by the Slovenian Research Agency (ARRS) through the Research program (P4-0220). The authors thank the editor and anonymous reviewers for their constructive comments, which helped us to improve the article.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.