
Abstract
Abstract
The female Aedes aegypti mosquito is an important vector for several tropical and subtropical diseases such as dengue, chikungunya, and Zika and yellow fever. The disease viruses infect the mosquito and subsequently spread to the salivary glands after which the viruses can be transmitted to humans with probing or feeding by the mosquito. Omics systems sciences offer the opportunity to characterize vectors and can inform disease surveillance, vector control and development of innovative diagnostics, personalized medicines, vaccines, and insecticide targets. Using high-resolution mass spectrometry, we performed an analysis of the A. aegypti salivary gland proteome. The A. aegypti proteome resulted in acquisition of 83,836 spectra. Upon searches against the protein database of the A. aegypti, these spectra were assigned to 5417 unique peptides, belonging to 1208 proteins. To the best of our knowledge, this is the largest set of proteins identified in the A. aegypti salivary gland. Of note, 29 proteins were involved in immunity-related pathways in salivary glands. A subset of these proteins is known to interact with disease viruses. Another 15 proteins with signal cleavage site were found to be secretory in nature, and thus possibly playing critical roles in blood meal ingestion. These findings provide a baseline to advance our understanding of vector-borne diseases and vector–pathogen interactions before virus transmission in global health, and might therefore enable future design and development of virus-blocking strategies and novel molecular targets in the mosquito vector A. aegypti.
Introduction
A
The incidence of dengue has increased dramatically in many countries in recent decades. Over 2.5 billion people are at risk of contracting dengue (WHO, 2015; Zelensky and Gready, 2005). Recently, the annual incidence of dengue has been estimated between 50 and 100 million infections worldwide. An estimated 500,000 people with severe dengue require hospitalization every year, a large proportion of these are children and about 2.5% of those affected die (WHO, 2015).
With recent surge in dengue in South East Asian countries and outbreak of Zika virus in Brazil and neighboring countries, the focus of management of these diseases has shifted heavily on vector control. Development of insecticide resistance in A. aegypti in South East Asia (Jirakanjanakit et al., 2007), Latin America (Rodriguez et al., 2005), and the Caribbean (Rawlins et al., 1998) is causing serious impediment in control of this medically important mosquito species. Several novel tools currently under different phases of development and evaluation have rekindled hope for the containment of A. aegypti in recent years.
Salivary glands of mosquitoes play an important role in blood meal acquisition and virus maintenance before their transmission. Saliva of the hematophagous arthropods contain angiogenic, anti-inflammatory, anticoagulant, and vasodilatory properties, which enable adult female uninterrupted blood feeding (Champagne and Ribeiro, 1994; Champagne et al., 1995; Ribeiro, 1992; Ribeiro et al., 1984; Stark and James, 1998). A 387 kDa protein in A. aegypti saliva has been shown to have immunomodulatory function, which lowers cytokine release and proliferation of T and B cells (Wasserman et al., 2004). Four proteins of 77, 58, 54, and 37 kDa found in the salivary gland extract from A. aegypti, with an ability to bind DENV serotypes, are likely to be involved in DENV infection of A. aegypti and Aedes polynesiensis. These proteins have been investigated for their utility as putative targets for transmission-blocking strategies (Cao-Lormeau, 2009). In addition, angiopoietin-like proteins have been found to be active in immunity-related responses (Wasinpiyamongkol et al., 2010). Interestingly, it has been demonstrated that overexpression of odorant binding proteins (OBPs) 10 and 22 (AAEL007603 and AAEL005772) were induced by virus, which resulted in enhanced host-seeking and probing behavior (Sim et al., 2012). There is a renewed interest in salivary constituents as potential biomarkers of exposure to vector bites, which can be useful in exposure surveillance (Fontaine et al., 2011; Remoue et al., 2006; Valenzuela et al., 2002).
A proteomic analysis of saliva from A. aegypti female mosquitoes offers the promise to reveal the presence of secretory proteins in the saliva. However, the collection of saliva for proteomics is more labor-intensive and extremely difficult to extract without contamination of neighboring tissues, than dissection of entire salivary gland. In contrast, a proteomic analysis of intact salivary gland can aid in identification of these potential secretory proteins along with proteins belonging to other cell constituents.
In addition, transcriptomic analysis has been carried out to understand gene expression pattern in the salivary glands and in response toward pathogens (Bonizzoni et al., 2012; Sim et al., 2012; Valenzuela et al., 2002). Valenzuela et al. (2002) have performed Edman degradation-based sequencing of protein bands obtained from salivary gland homogenates and identified 11 proteins. In a subsequent study, expression of 24 proteins has been described from the salivary glands of A. aegypti (Ribeiro et al., 2007). In a recent study, 13 immunogenic proteins have been identified from 13 and 56 kDa protein bands from the salivary gland proteome of A. aegypti (Oktarianti et al., 2015). Among these, the D7 arthropod OBP and apyrase were most abundant in 31 and 56 kDa bands, respectively. Mass spectrometry-based proteomic analysis of salivary glands of female Anopheles gambiae mosquito has been employed by independent studies (Chaerkady et al., 2011; Kalume et al., 2005). However, a global proteomic profiling of salivary glands of A. aegypti is not yet available.
We carried out an unbiased proteomic analysis of A. aegypti salivary glands using high-resolution LTQ-Orbitrap Velos mass spectrometer. We believe that the salivary gland proteome described in this study could pave way to devise innovative strategies against transmission dengue and some of the other mosquito-borne diseases.
Materials and Methods
Rearing and establishment of the A. aegypti colony
A. aegypti was sourced from the insectary of National Institute of Malaria Research, field unit at Goa. In this facility, the larvae were reared in plastic trays containing tap water at 27°C ± 2°C, relative humidity of 70% ± 5%, and a photoperiod: scotoperiod of 12:12 h (light: dark cycles). The larvae were fed on Cerelac™ powder till their metamorphosis into pupae. The latter were kept in 300 mL plastic bowls inside the cloth cages for emergence of adults. Glucose solution (10%) soaked in cotton pad was provided to the adults. After mating, the females were fed on blood using standard protocols. Plastic bowls of 300 mL capacity containing water and lined with filter papers were placed in the cage for facilitating oviposition. In this manner, a continuous cyclic colony of the species was maintained in the insectary. This study has the approval of institutional ethics committee of NIMR, ICMR, New Delhi.
Salivary gland dissection of female A. aegypti
Glucose-fed adult female A. aegypti were dissected for salivary glands (Fig. 1a). The dissection was carried out in 0.65% normal saline under dissecting microscope. The dissected salivary glands were washed in saline twice and were immediately transferred to the PBS buffer and stored at −80°C.
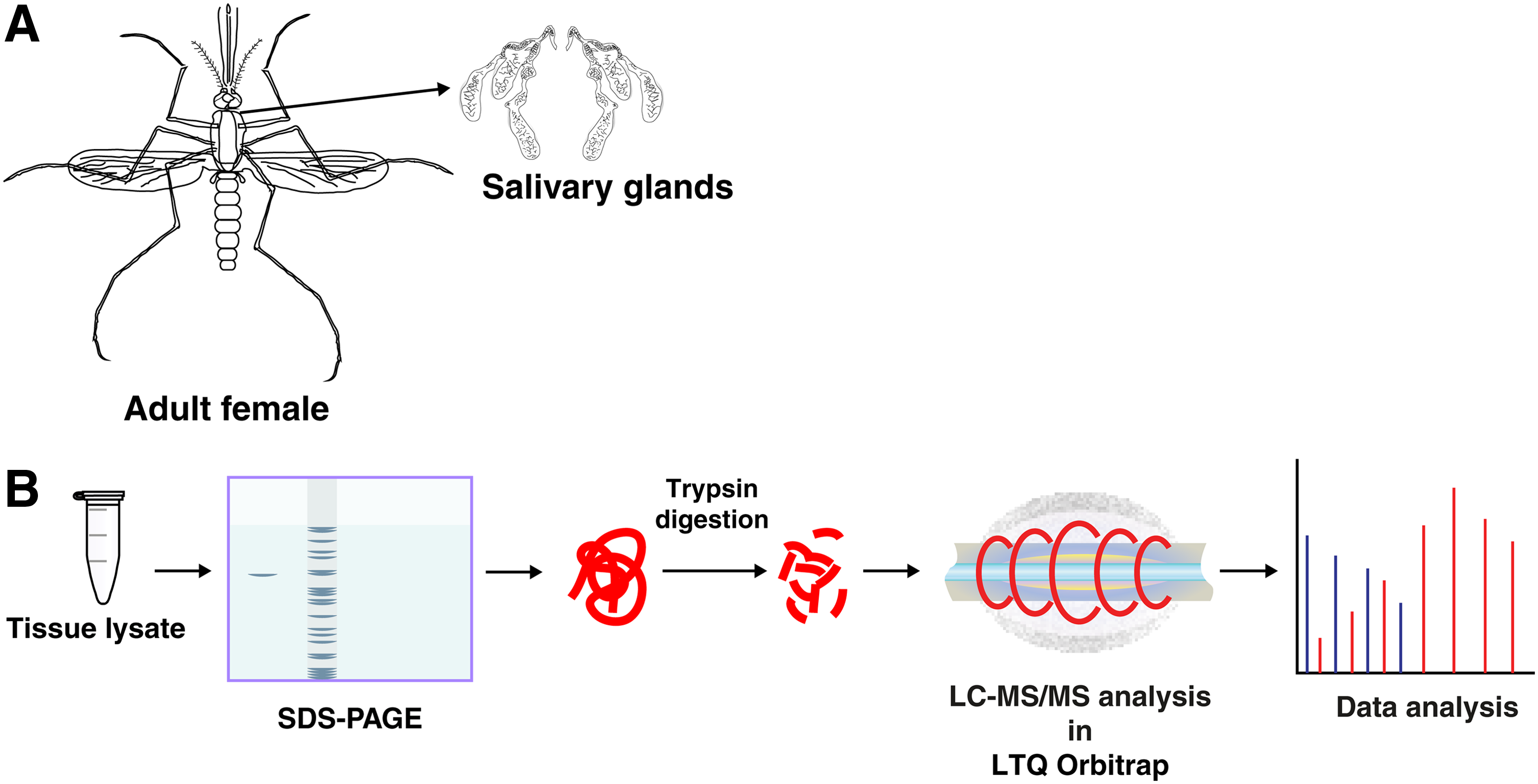
Protein extraction from salivary glands
In all, 500 salivary glands were homogenized in 200 μL of 0.5% SDS by ultrasonication. The ultrasonication was performed using Branson Sonifier SFX150 at amplitude of 40%. The sonicator was on for 10 sec, off for 20 sec, and the same sonication cycle was applied for 10 times. The tube was kept on ice during sonication to avoid generation of heat. This suspension was then centrifuged for 20 min at 14,000 rpm. The supernatant was collected and protein amount was estimated using Bicinchoninic acid assay (Pierce®; Cat. No. 23225). The SDS-PAGE profile of the protein lysate is shown in Supplementary Figure S1.
In-gel trypsin digestion of proteins
Two hundred micrograms of protein was resolved on SDS-PAGE, stained with colloidal Coomassie blue, and destained gel bands were processed for in-gel digestion as previously described (Prasad et al., 2016). The destained gel bands were subjected to reduction by 5 mM of dithiothreitol followed by alkylation with 10 mM of iodoacetamide. The in-gel digestion of proteins was carried out using trypsin in a trypsin:sample ratio of 1:20 (modified sequencing grade; Promega, Madison, WI) at 37°C for 16 h. Digested peptides were then extracted and stored at −80°C until LC-MS/MS analysis (Fig. 1b).
LC-MS/MS analysis
LC-MS/MS analysis of the in-gel fractions was carried out on LTQ–Orbitrap Velos (Thermo, Bremen, Germany) connected with Proxeon Easy nLC system (Thermo Scientific, Bremen, Germany). Peptides were first enriched on a trap column (75 μm × 2 cm) packed in-house using C18 material (Magic C18AQ, 5 μm, 100 Å, Michrom Biosciences, Inc.) with a flow rate of 3 μL/min and resolved on an analytical column (75 μm × 10 cm, Magic C18AQ, 3 μm, 100 Å; Michrom Biosciences, Inc.) at a flow rate of 350 nL/min using a linear gradient of 7–30% acetonitrile over 70 min. The mass spectrometry analysis was carried out in a data-dependent analysis mode with survey scans (MS) acquired at a resolution of 60,000 at m/z 400 and fragment ion scan (MS/MS) acquired at a resolution of 15,000 at m/z 400. Both MS and MS/MS scans were acquired in Orbitrap mass analyzer. Precursor ions were fragmented by high collision-induced dissociation (normalized collision energy value of 39%). The parameter settings for LTQ–Orbitrap Velos analysis were as follows: (a) up to 20 MS/MS scans per duty cycle; (b) precursor ions were dynamically excluded for a period of 45 sec; (c) for MS/MS analysis, monoisotopic precursor mass selection and rejection of singly charged ion criteria were enabled; (d) capillary temperature was set at 250°C; and (e) the automatic gain control was set at 1 × 106 for MS and 1 × 105 for MS/MS with maximum time for accumulation of 100 for MS and 250 for MS/MS scans. The lock mass option was enabled and polydimethylcyclosiloxane (m/z, 445.120025) was used for internal calibration.
Database searches for protein identification
High-resolution MS/MS spectra were searched against the protein database of A. aegypti using Sequest and Mascot search engines incorporated with the Proteome Discoverer software suite (Thermo Fischer Scientific, Bremen, Germany). The search parameters used were as follows: (a) trypsin as the proteolytic enzyme (with up to one missed cleavage); (b) peptide mass error tolerance of 20 ppm; (c) fragment mass error tolerance of 0.1 Da; (d) carbamidomethylation of cysteine as fixed modification; and (e) oxidation of methionine as a variable modification. A false discovery rate of 1% was applied while identifying the peptide–spectrum matches (PSMs).
Bioinformatics analysis
The bioinformatics analysis of protein data was carried out using VectorBase resources for assigning gene ontology (GO) terms. KEGG pathway portals were used to assess the involvement of proteins in different metabolic pathways. Among the datasets, immunogenic proteins were identified using ImmunoDB (http://cegg.unige.ch/Insecta/immunodb/), predictive functional analysis of proteins was carried out using NCBI Blastp by blast2go search engine, and protein–protein interaction networks were mapped using STRING v 9.1 (Franceschini et al., 2013).
Results and Discussion
LC-MS/MS analysis of 18 fractions of A. aegypti salivary gland proteins processed on high-resolution mass spectrometer resulted in the acquisition of 83,836 MS/MS spectra. These MS/MS spectra when searched against protein database of A. aegypti led to identification of 5417 unique peptides and 1208 salivary gland proteins (Supplementary Table S1).
Molecular functions were assigned to these proteins using VectorBase resource and GO (Giraldo-Calderón et al., 2015). The most prominent biological processes represented in our data were found to be translation (15%), metabolism (11%), oxidation-reduction (9%), and cellular organization (8%). At least 5% of the proteins were found to be involved in signal transduction and metabolic pathways (Fig. 2). Majority of these proteins were found to be belonging to membrane (18%), intracellular (13%), ribosomal (12%), and cytoplasmic (10%) regions (Fig. 2).
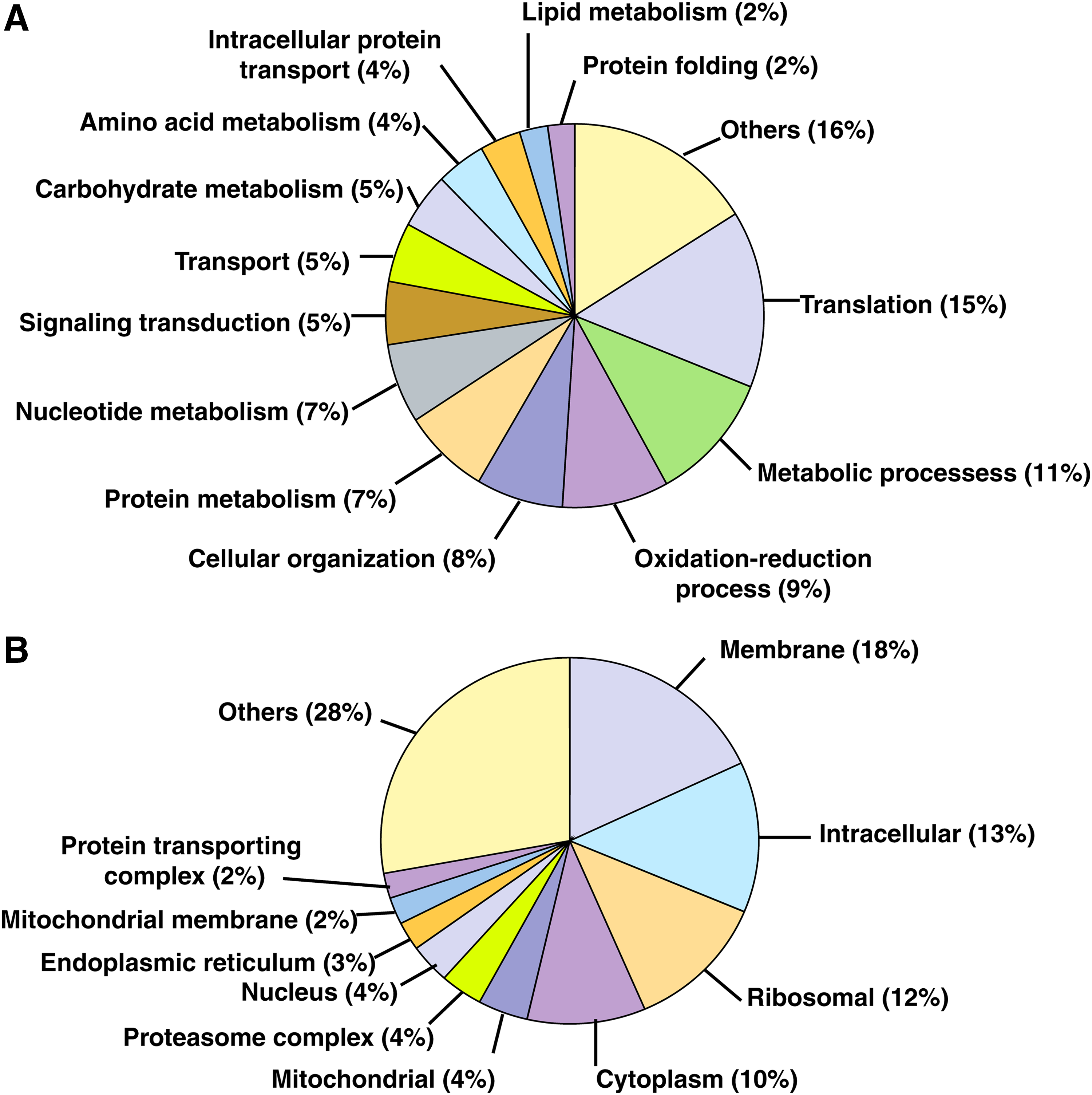
Subcellular localization and functional annotation of identified proteins.
Among all the proteins, 238 could not be ascribed any GO assignment by VectorBase (Supplementary Table S2). However, 64 of these were predicted to have a SignalP cleavage site and are likely to be secretory in nature. Another 10 proteins were assigned with an ImmunoDB identity. These include CTL-16, CTL-25, CTLGA2, CTLGA3, FREP-20, FREP-22, GAM, LOQS, ML1, and SRPN 26. Out of a total of 1208 proteins identified, 287 (23.8%) were listed as hypothetical proteins in Vector Base. Of these, 150 hypothetical proteins have been assigned a GO term. However, 137 proteins are yet to be functionally characterized and ascribed a GO term. A predictive functional analysis of these 137 protein sequences was carried out by NCBI Blastp through blast2go (Götz et al., 2011) tool against a nonredundant database. Orthologs of the sequence were identified based on top blast hits, and predictive GO domains could be then assigned to 122 out of these 137 hypothetical proteins (Supplementary Table S3).
We compared our protein data with proteins previously identified to be involved in interaction with dengue virus. For example, endoplasmin (AAEL012827), a heat shock protein in endoplasmic reticulum also known as gp96, was identified with 36 unique peptides in our study. Endoplasmin is known to interact with dengue virus proteins NS3 and NS5. It possesses a signalP cleavage site, which indicates its probable secretory function. It was speculated to be the initial step in the initiation of innate immune response to dengue infection as a chaperon required for Toll receptor folding (Zelensky and Gready, 2005). A recent study has demonstrated the inhibitory role of salivary gland extract in dengue virus transmission to vertebrates, which implicated direct interaction of D7 proteins with the virions (Conway et al., 2016). Another study reported 94 proteins in multiple larval stages and adult A. aegypti, which directly interact with different DENV proteins (Mairiang et al., 2013). Of these 94 proteins, 24 were identified in our dataset, confirming their expression in salivary gland (Table 1).
We identified two OBPs in our study, namely OBP-22 (AAEL005772) and OBP-17 (AAEL004339). OBPs form a part of chemosensory apparatus in insects. In an RNAi-mediated gene knockdown experiment, OBP-22 has been found to be upregulated in DENV-infected salivary glands of A. aegypti. It has been shown to facilitate host-seeking and probing behavior and thus may affect the transmission efficiency (Sim et al., 2012). Identification of such proteins may provide novel insights into the molecular mechanism of host-seeking behavior and disease transmission by A. aegypti.
Protein–protein interactions in metabolic pathways
Out of the total 1208 proteins identified in this study, 402 (33.27%) proteins were found to be involved in one or more metabolic pathways as described for A. aegypti in KEGG Pathways (Sumimoto, 2008) (Supplementary Table S4). As expected, majority of the proteins were found involved in house-keeping pathways, including glycolysis, TCA cycle, fatty acid metabolism, amino acid metabolism, biosynthesis of antibiotics, glyoxylate and dicarboxylate metabolism, and carbon metabolism.
Apart from these, proteins from the WNT signaling pathway, Hedgehog, TGF-beta, Hippo, FoxO, mTOR, and peroxisome signaling pathways were also identified, signifying their functional presence in salivary glands (Supplementary Table S4). We identified 15 out of 69 known members of peroxisome pathway in our dataset (Table 2). The members of this pathway have been shown to be differentially expressed in response to dengue virus infection (Götz et al., 2011). Among these, four immunogenic proteins, including the two known isoforms of Catalase 1B, Cu Superoxide Dismutase2, and Mn Superoxide Dismutase1 (AAEL013407-RB, AAEL013407-RA, AAEL006271-RC, and AAEL004823-RA), were identified in this study (Fig. 3a). On using the Markov Cluster algorithm (MCL), three well-defined clusters were marked within the pathway (Fig. 3b). The clustering pattern indicates close functional association of catalases with several superoxide dismutases, which are well known for their role in regulating the reactive oxygen species (ROS) in insects. ROS has also been implicated in innate immunity in insects contributing to apoptosis, necrosis, mitogenesis, host defense, and gene expression (Sumimoto, 2008).

Immunogenic salivary proteins
Among all the proteins identified, 29 proteins were found to be enlisted in ImmunoDB dataset for A. aegypti, implying their evidenced role in immunity. These immunogenic proteins included members of 1,3-beta-
A. aegypti genome has seven GNBPs classified into GNBPA and GNBPB. GNBPB subfamily is unique to mosquitoes. A. aegypti GNBPs have shown response when challenged by Escherichia coli, Staphylococcus aureus, and Beauveria bassiana (Sumimoto, 2008). GNBPs have been shown to be involved in initial prophenol oxidase (PPO) cascade activation. GNBPs are pattern recognition receptors known to counter Plasmodium infection and participate in innate immune response in A. gambiae (Beutler, 2004). They not only regulate immune signaling pathways but also interact with Gram-negative E. coli. GNBPB1 in A. gambiae has been indicated with resistance to Plasmodium berghei and E. coli infection (Beutler, 2004). We identified six unique peptides against GNBPB6 in salivary gland.
In contrast to A. gambiae, where only one isoform has been reported for catalase gene, two isoforms are known (AAEL013407-RA and RB) in A. aegypti. Both of these isoforms were identified in this study. Catalases have been found to be involved in at least four signaling pathways—glycoxylate and dicarboxylate metabolism, tryptophan metabolism, and peroxisome pathway. Catalases get activated in response to oxidative stress and are involved in catabolic processes mediated by hydrogen peroxide (H2O2). At the metabolic level, catalases play additional roles in heme and metal ion binding.
Similarly, superoxide dismutases are known to be involved in antiviral response. Both the Cu and Mn superoxide dismutases (AAEL006271, AAEL004823) identified in this study were predicted to be secretory in nature and are known to be involved in the peroxisome pathway. In addition, we found glutathione peroxidase (GPXH1) (AAEL012069-RB), 5 thioredoxin peroxidases (TPX 1–5) (AAEL013528, AAEL004112, AAEL014548, AAEL002309, and AAEL009051), peroxiredoxin (AAEL007135-RA), and glutathione S-transferase (GST) (AAEL006829-RA). These are the primary antioxidant enzymes that act directly on ROS. In addition to these, we also identified a dual oxidase (DUOX) (AAEL007563-RA). DUOX has been described in Drosophila melanogaster and A. gambiae to be producing ROS and also playing a role in host defense and hormone production (Beutler, 2004; Iwanaga and Lee, 2005).
We identified a clip-domain serine protease protein with multiple PSMs. The gene family of CLIP proteases is known to regulate immune responses in insects (Jiang and Kanost, 2000). The CLIP proteases in hemolymph get stimulated by infections and activate the PPOs or Toll Ligand pathway. The functions of a very few CLIP proteases have been described as yet and more experimental studies would be necessary to understand their role in immune responses in insects. The role of CLIP in Drosophila has been described in the antimicrobial peptide (AMP) producing Toll pathway and PPO conversion to phenol oxidase, which then melanizes the parasites in insects (Jang et al., 2008). A reverse genetics approach has been recently used to identify the role of CLIP in melanization responses of A. gambiae to P. berghei (murine malaria) (Barillas-Mury, 2007). Mosquito proteins involved in the melanization processes have been reviewed recently (Sreenivasamurthy et al., 2013). CTLs are among such proteins and four of them were identified in this study. These proteins are CTL16 (AAEL000533), CTL25 (AAEL000556), CTLGA2 (AAEL013566), and CTLGA3 (AAEL011070). All of them have a signal domain indicating their secretory function. CTLs are calcium-dependent proteins, which in invertebrates mediate immune responses, including opsonization, activation of PPO causing melanization, and microbial clearance (Zelensky and Gready, 2005). In a recent transcript study of sialome of male and female A. aegypti, CTLs were overtranscribed many folds in female glands. This study attributes it to blood feeding by female mosquito and role of CTLs in immune modulation (Ribeiro et al., 2016).
Multiple peptide evidences against fibrinogen-related proteins FREP 20 (AAEL000726) and FREP 22 (AAEL000749) proteins were obtained in this study. Both these proteins possess signal peptide domain and are likely to be secreted. FREP 22 has known antibacterial properties and a role in innate immune response (Thangamani and Wikel, 2009). FREPs belong to a well-known and largest pattern recognition receptor immune gene family, also called as fibrinogen-domain immune-lectins (FBN). It has already been demonstrated that FREPs have sex- and tissue-specific responses (Chang et al., 2005; Cirimotich et al., 2010; Dong and Dimopoulos, 2009; Fan et al., 2008; Hanington and Zhang, 2011; Jiravanichpaisal et al., 2006; Kaneko et al., 2006; Kuo et al., 2006; Middha and Wang, 2008). In the past, FBN22 has been shown to be induced in response to infection by both Gram-negative and Gram-positive bacteria (Fujita, 2002; Lu et al., 2002; Matsushita and Fujita, 2002; Teh et al., 2000). Thus, it appears that the role of FREPs in salivary gland may well be predominantly toward innate immune response with anti-Plasmodium and antibacterial activities. Gambicin, a cysteine-rich AMP, has been identified only in A. gambiae and A. aegypti so far. It has an activity against Gram-positive and Gram-negative bacteria and the ookinete of malaria parasite. It is known for its expression in anterior midgut, abdomen, and thorax (Vizioli et al., 2001).
Conclusions and Future Outlook
In this study, we have employed mass spectrometry-based approach to understand salivary gland proteome of female A. aegypti. To the best of our understanding, this is so far the largest catalogue of salivary gland proteome of A. aegypti. Salivary gland proteins identified to be secretory are likely to play a significant role in blood ingestion and disease transmission. Humans develop antisalivary proteins after exposure to such secreted proteins upon mosquito bite. These antisalivary proteins can be targeted as epidemiological markers for exposure to the vector. In addition, subsets of the proteins identified to be involved in mosquito immunity and direct interaction with the viral proteins are attractive candidates for the development of transmission-blocking vaccines.
Several interesting hypotheses can be considered based on our study findings, its analyses, and interpretations. However, there is a need to carry out deeper functional studies on the proteins identified in this study, which remains the limitation of our study in its current form. A deeper understanding based on investigation into the functioning of salivary glands as immune-modulatory organs may render them as primary transmission-blocking sites and for the implementation of targeted interventions against disease pathogens.
Involvement of proteins in immune-related metabolic pathways in salivary glands can be a valid ground for further studies in understanding the transmission physiology of disease-causing viruses. The rich protein–protein interaction networks operating in the salivary glands further points to the need for identification of critical points in the metabolic pathways. A comparative proteomic analysis of salivary gland upon infected blood meal will allow comparison of differential gene expression in response to the infection. Such analysis will reveal the proteins involved in blood meal metabolism and immunity against the viral infection.
The proteomic data generated in this study will serve as a baseline to further understanding of molecular-level vector–pathogen interactions before virus transmission, and might enable future design and development of virus-blocking strategies in the mosquito vector A. aegypti.
Footnotes
Acknowledgments
We thank the Indian Council of Medical Research, National Institute of Malaria Research, Institute of Bioinformatics, and Goa University for institutional support. This article bears the NIMR publication screening committee approval no. 028/2016. We are grateful to Dr. Neena Valecha, Director NIMR, for encouragement. We thank Ministry of Defence and Armed Forces Medical College (AFMC) for research approval to R.D. We thank the Department of Biotechnology (DBT), Government of India, and Infosys Foundation for research support to the Institute of Bioinformatics, the Department of Health and Family Welfare, for procuring mass spectrometer at NIMHANS-IOB laboratory, the Science and Engineering Research Board (SERB), Government of India, for funding (EMR/2014/000444) to NIMR and IOB to characterize proteome and transcriptome of mosquito vectors, the Council of Scientific and Industrial Research, Government of India, for research fellowships to M.K. and J.A, and the University Grants Commission, Government of India, for research fellowships to G.D. IOB is also supported by DBT Program Support on infrastructure for proteomic data analysis (BT/01/COE/08/05).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.