
Abstract
Abstract
Ophthalmology and visual health research have received relatively limited attention from the personalized medicine community, but this trend is rapidly changing. Postgenomics technologies such as proteomics are being utilized to establish a baseline biological variation map of the human eye and related tissues. In this context, the choroid is the vascular layer situated between the outer sclera and the inner retina. The choroidal circulation serves the photoreceptors and retinal pigment epithelium (RPE). The RPE is a layer of cuboidal epithelial cells adjacent to the neurosensory retina and maintains the outer limit of the blood–retina barrier. Abnormal changes in choroid–RPE layers have been associated with age-related macular degeneration. We report here the proteome of the healthy human choroid–RPE complex, using reverse phase liquid chromatography and mass spectrometry-based proteomics. A total of 5309 nonredundant proteins were identified. Functional analysis of the identified proteins further pointed to molecular targets related to protein metabolism, regulation of nucleic acid metabolism, transport, cell growth, and/or maintenance and immune response. The top canonical pathways in which the choroid proteins participated were integrin signaling, mitochondrial dysfunction, regulation of eIF4 and p70S6K signaling, and clathrin-mediated endocytosis signaling. This study illustrates the largest number of proteins identified in human choroid–RPE complex to date and might serve as a valuable resource for future investigations and biomarker discovery in support of postgenomics ophthalmology and precision medicine.
Introduction
O
The RPE is a monolayer of pigmented cuboidal cells situated between photoreceptors and Bruch's membrane (Camelo et al., 2015). Anatomically, RPE is close to the outer segment of the photoreceptors at its apical surface (Lamb and Pugh, 2004). The hexanocuboidal layer of cells of RPE is crucial for the maintenance and survival of the overlying photoreceptor cells and is also found to regulate the integrity of the choroidal capillaries (Boulton and Dayhaw-Barker, 2001). The RPE through its junctional complexes constitutes the outer blood–retina barrier.
Its other functions include transport of ions, water, and metabolic end products from the subretinal space to the blood, phagocytosis of the shed outer segments of photoreceptors, and directional transport of nutrients (Bonilha et al., 2006). Changes in the choroid are noticed in various inflammatory conditions such as sympathetic ophthalmia, Vogt–Koyanagi–Harada syndrome, sarcoidosis, tuberculosis, toxoplasmosis, and syphilis. RPE dysfunction is central to the pathophysiology of age-related macular degeneration (AMD), central serous chorioretinopathy, and various hereditary retinal degenerations (Bhutto and Lutty, 2012).
Until recently, mass spectrometry (MS)-based proteomics was limited to partial proteome analysis. Presently with the availability of more sensitive and powerful mass spectrometers, in-depth coverage of proteomes of mammalian cells including the various parts of the human eye is possible (Goel et al., 2013; Kim et al., 2014; Mann et al., 2013). In recent years, several proteomic studies have been carried out to understand the proteome of the various tissues of the healthy human eye (Crabb, 2014; Murthy et al., 2014; Skeie and Mahajan, 2014).
Similarly, attempts have been made to identify biomarkers in eye-related diseases (Koss et al., 2014; Priyadarsini et al., 2014). In this study, we used a MS-based proteomic approach to analyze the proteome of human choroid–RPE tissue harvested from three cadavers and identified 5309 proteins. A large majority of these proteins were found to be associated with a role in transport of ions, metabolism, cell growth and maintenance, thermoregulation, and secretion of growth factors. To the best of our knowledge, this study reports the largest number of proteins from choroid of human eye. The information derived from this study could serve as a platform for future proteomic studies on the choroid–RPE complex.
Materials and Methods
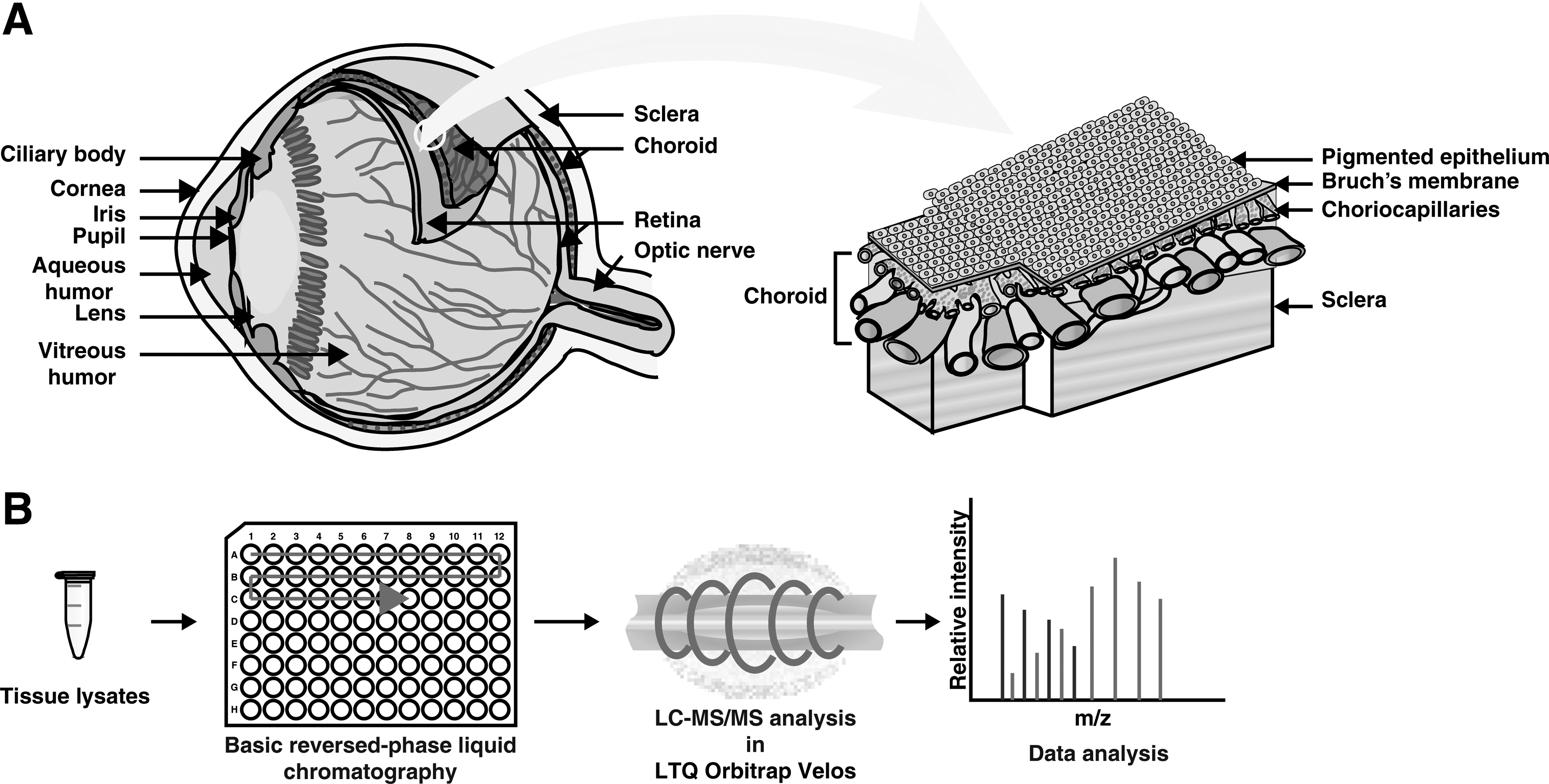
Sample collection
The choroid–RPE tissues were acquired from healthy donor eyes after obtaining approval from the institutional review board of Vittala International Institute of Ophthalmology, Bangalore, India. The study had institutional ethic board approval and adhered to the tenets of declaration of Helsinki. The choroid–RPE tissue was dissected from three healthy donor eyes, which were enucleated within 6 h of death. During the dissection of the eye ball, care was taken to make sure that there was no retinal tissue contamination. After dissecting out the cornea, iris, and lens, a sclera window was created near the equator, exposing the choroid. A nick was created in the choroid to gain entry into the subretinal space. Hydrodissection of the retina from the choroid–RPE complex was performed by injecting a fluid wave into the subretinal space. The retinal tissue and the choroid–RPE complex were then separately harvested. The collected specimens were frozen and stored at −80°C until further analysis. The details of the tissues used are provided in the Supplementary Table S1.
Sample preparation and fractionation
The choroid–RPE tissue was lysed using buffer containing 4% sodium dodecyl sulfate (SDS), 100 mM dithiothreitol, and 100 mM Tris; pH 7.5. The tissue lysates were then homogenized and sonicated followed by heating for 10–15 min at 75°C. The lysates were cooled, centrifuged at 10,000 rpm for 10 min, and buffer exchange was carried out. The protein concentration of the cleared lysate was estimated using bicinchoninic acid assay. For further analysis, an equal amount of protein from each of the donor tissue was pooled. Filter-aided sample preparation (Wisniewski et al., 2009) was carried out.
In brief, 450 μg of proteins from the pooled samples was buffer exchanged with 8 M urea in a 30 kDa MWCO spin filters (EMD Millipore, Billerica, MA, USA) to reduce the SDS concentration. This was followed by alkylation of cysteine residues with 10 mM iodoacetamide. The proteins were digested using sequencing-grade modified trypsin (Cat. No. V5111; Promega, Madison, WI, USA) overnight at 37°C. The peptide digest was desalted using Sep-Pak C18 columns (Waters Corporation, Milford, MA, USA) and lyophilized.
Before high pH reverse phase chromatography fractionation, the lyophilized samples were reconstituted in basic reverse phase liquid chromatography (bRPLC) solvent A (10 mM triethyl ammonium bicarbonate [TEABC], pH 8.5) and loaded on XBridge C18, 5 μm 250 × 4.6 mm column (Waters, Milford, MA, USA) connected to Agilent 1100 series high-performance liquid chromatography system. Using a gradient of 0–100% solvent B (10 mM TEABC in acetonitrile, pH 8.5), the peptide digest was resolved in 50 min. A total of 96 fractions were collected, which were pooled to 24 fractions, vacuum dried, and stored at −80°C until further liquid chromatography-tandem mass spectrometry (LC-MS) analysis.
LC-MS/MS analysis
The bRPLC-fractionated peptide digests were analyzed on LTQ-OrbitrapVelos mass spectrometer (Thermo Electron, Bremen, Germany) interfaced with Easy-nLC II nanoflow liquid chromatography system (Thermo Scientific, Odense, Southern Denmark). The peptide digests from each fraction were reconstituted in solvent A (0.1% formic acid) and loaded onto trap column (75 μm × 2 cm) packed in-house with Magic C18 AQ (MichromBioresources, Inc., Auburn, CA, USA) (5 μm particle size, pore size 100 Å) at a flow rate of 5 μL/min with solvent A (0.1% formic acid in water). Peptides were resolved on an analytical column (75 μm × 15 cm) at a flow rate of 300 μL/min using a linear gradient of 7–30% solvent B (0.1% formic acid in 95% acetonitrile) over 60 min.
MS analysis was carried out in a data-dependent manner. The precursor scans (MS data) were acquired in the Orbitrap mass analyzer at a mass resolution of 60,000 at 400 m/z in the m/z scan range of 350–1800. Eight most intense precursor ions from a survey scan were selected for MS/MS from each duty cycle and detected at a mass resolution of 15,000 at m/z of 400 in the Orbitrap analyzer. The fragmentation was carried out using higher energy collision dissociation with 35% normalized collision energy. Dynamic exclusion was set for 30 sec with a 10 ppm mass window. Internal calibration was done using lock mass from ambient air (m/z 445.1200025) (Wisniewski et al., 2009).
Data analysis
MS-derived data were searched against National Center for Biotechnology Information Human RefSeq70 protein database (along with common contaminants) using Sequest and Mascot (version 2.2) search algorithms through the Proteome Discoverer 2.0 software suite (Thermo Scientific, Bremen, Germany). Carbamidomethylation of cysteine was specified as fixed modification, and acetylation of protein N-termini and oxidation of methionine were included as variable modifications. Trypsin was specified as protease with maximum of two missed cleavage allowed. The precursor mass tolerance and the fragment mass tolerance were set to 10 ppm and 0.05 Da, respectively. The data were also searched against decoy database and the false discovery rate was set to 1% at the peptide level.
Bioinformatics analysis
The proteins identified in this study were categorized based on their primary subcellular localization, molecular function, and biological process annotations in Human Protein Reference Database (HPRD; www.hprd.org), which is a gene ontology compliant database (Muthusamy et al., 2013; Prasad et al., 2009). These proteins were also compared with the data available through Human Proteome Map portal (www.humanproteomemap.org) (Kim et al., 2014) and neXtProt missing proteome data (www.nextprot.org/db/) (Gaudet et al., 2013). Furthermore, choroidal proteins were subjected to functional analyses using QIAGEN's Ingenuity Pathway Analysis (IPA; www.qiagen.com/ingenuity) to assess the protein networks enriched in the human choroid–RPE complex tissue.
Data availability
The MS-derived data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the ProteomicsIDEntifications database partner repository. The data set identifier is PXD002273.
Results and Discussion
In this study, we carried out an in-depth analysis of the human choroid–RPE complex tissue proteome by employing protein and peptide-fractionation methods followed by LC-MS/MS. The analysis of 289,226 mass spectra resulted in the identification of 26,503 unique peptides from 5309 proteins. This proteomic data set categorizes novel molecular signatures in anatomically sensitive regions of the choroid–RPE complex. The findings give systematic insight into choroid–RPE function, reveal significant choroid–RPE processes, and prioritize new pathways for future targeted research. The major biological functions of choroid–RPE proteins include protein metabolism, regulation of nucleic acid metabolism, transport, cell growth, and/or maintenance and immune response.
The experimental strategy used to characterize the human choroid–RPE complex is illustrated in Figure 1. A complete list of proteins identified in the human choroid–RPE complex and intensity-based absolute quantification (iBAQ) values are provided in Supplementary Table S2. This table describes the functional analysis of the proteins and iBAQ values in detail. A list of nonredundant peptides identified from this study is given in Supplementary Table S3.
Despite recent efforts by various research groups to profile the proteome of the various parts of human eye, choroid–RPE proteome remains less studied. The only previously published study (Skeie and Mahajan, 2014) has used choroid–RPE complex collected from three donors of age between 80 and 90 years and reported the identification of 4403 unique proteins. We identified 5309 proteins from choroid–RPE in this study. Functional analysis of these proteins included previously identified risk factors for retinal diseases related to inflammation, oxidative stress, and the complement cascade. IPA of these unique molecules found to be associated with the network functions related to inflammatory diseases, metabolism, molecular transport, cellular function, and maintenance. These network functions support the familiarity of choroid–RPE functions. Hence the findings of this study further enhanced the biochemical understanding of choroid–RPE by adding potential roles of 909 proteins.
Summary of proteomics data
Two of the previously reported proteins in this study is discussed in the following subsection and their representative mass spectra are shown in Figure 2A and 2B.
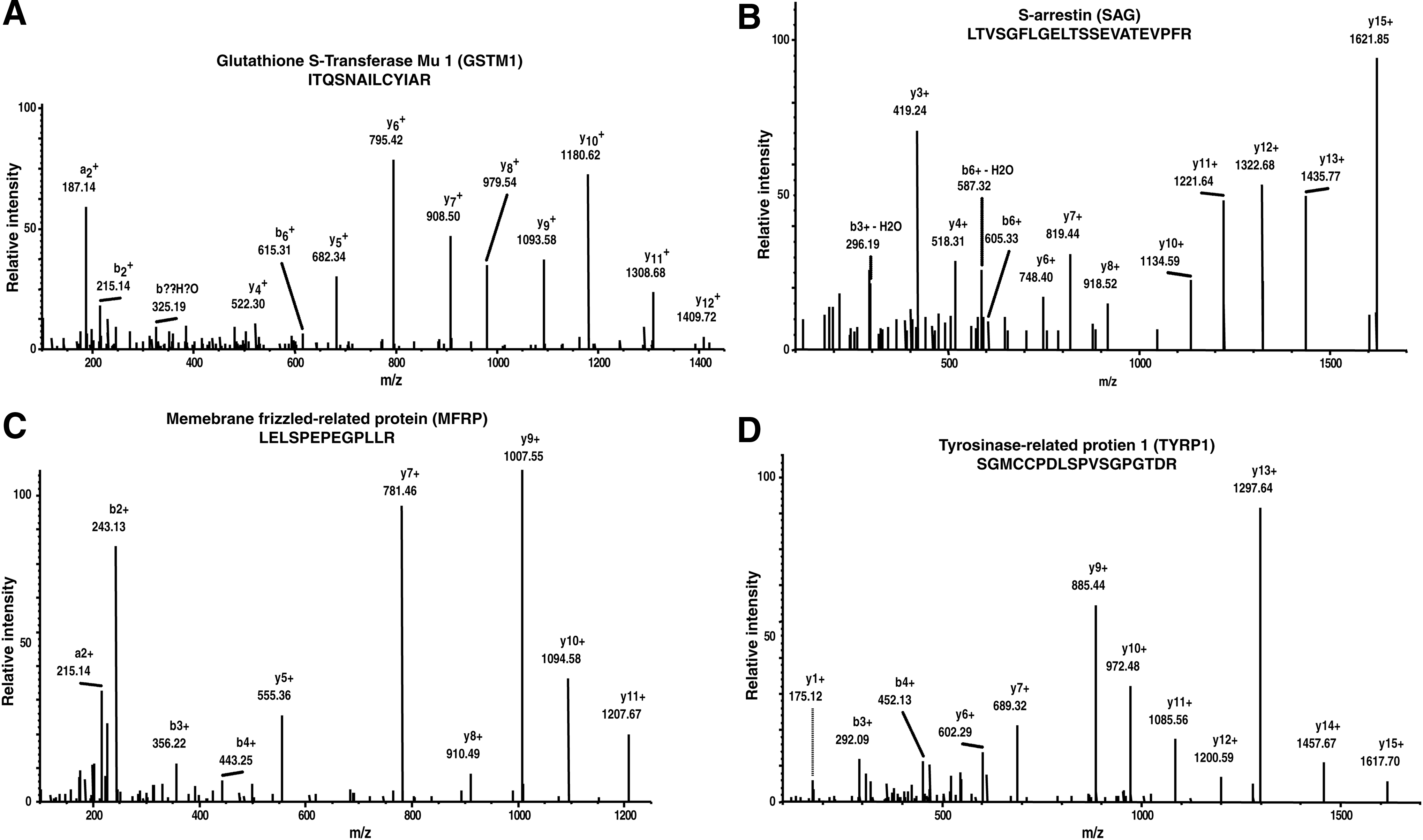
Representative MS/MS spectra of previously reported proteins.
Glutathione S-transferase Mu 1
Cytosolic and membrane-bound forms of glutathione S-transferase (GST) are encoded by two distinct supergene families. The mammalian cytosolic GSTs have been grouped into eight classes, which are designated alpha, kappa, mu, omega, pi, sigma, theta, and zeta. This gene encodes a GST that belongs to the mu class and it is polymorphic in humans (To-Figueras et al., 1997). The mu class of enzymes plays a role in the detoxification of electrophilic compounds, including carcinogens, therapeutic drugs, environmental toxins, and products of oxidative stress, by conjugation with glutathione (Rossini et al., 2002). Polymorphisms of glutathione S-transferase Mu 1 (GSTM1) have previously been reported to be associated with glaucoma, cataract, exudative AMD, as well as various spontaneous optic neuropathies (Abu-Amero et al., 2009; Othman et al., 2012). It is also studied that a GSTM1-positive genotype was a risk factor for developing primary open angle glaucoma (Safa et al., 2014).
S-arrestin
Arrestin (retinal S-antigen, SAG) is a major protein of the retinal rod outer segments. It interacts with photo-activated phosphorylated rhodopsin, inhibiting or arresting its ability to interact with transducin. S-arrestin, also known as SAG, is a major soluble photoreceptor protein that is involved in desensitization of the photoactivated transduction cascade (Wilson and Applebury, 1993). It is highly antigenic, and is capable of inducing experimental autoimmune uveitis. Mutations in this gene have been associated with Oguchi disease, a rare autosomal recessive form of night blindness, retinitis pigmentosa, and retina degenerative diseases (Huang et al., 2012; Sonoyama et al., 2011).
Comparison with human proteome
We compared data obtained in this study with “A draft map of the human proteome” (Kim et al., 2014), and the comparison resulted in the identification of 5242 common proteins and 35 proteins unique to this study. Out of these 35 proteins, 5 proteins are identified with multiple peptide spectral matches (PSMs) and peptides (Table 1). Additional studies are essential to understand the role of these proteins in the functioning of choroid–RPE. Two of the important molecules are discussed in the following subsections and their representative spectra are shown in Figure 2C and 2D.
Membrane frizzled-related protein
This protein is a member of the frizzled-related protein family. Frizzled proteins are seven-transmembrane-type WNT (wingless-type) receptors that transduce WNT signals to the intracellular signaling pathways (Cong et al., 2004). Membrane frizzled-related protein (MFRP) has been found to be essential for normal development and maintenance of outer segments of photoreceptors (Won et al., 2008). MFRP has also been studied for its involvement in primary angle closure glaucoma and the development of nanophthalmos (Shi et al., 2013). Mutations in this protein coding gene have been associated with nanophthalmos (Sundin et al., 2005), posterior microphthalmia, retinitis pigmentosa, foveoschisis, and optic disk drusen (Paun et al., 2012; Zacharias et al., 2015).
Tyrosinase-related protein 1
This protein is encoded by a gene called 5,6-dihydroxyindole-2-carboxylic acid oxidase precursor (TYRP1). Tyrosinase-related protein 1 (TYRP1) is a ∼75 kDa molecular weight protein and appears to be the main abundant melanosomal protein of the melanocyte. TYRP1 contains 537 amino acid residues and shares 40–52% of amino acid homology to tyrosinase protein (Fang et al., 2002). TYRP1 is involved in the maintenance of melanosome structure and affects melanocyte proliferation and cell death (Kamaraj and Purohit, 2013). RPE melanin plays essential role in the development of the neural retina. In choroid–RPE, it may play a role in the maintenance of melanosome structure. It is also studied for its role in the regulation in pigmentation metabolism (Lu et al., 2011).
Comparison of choroid proteome with iris tissue and ciliary body
The choroid is considered as a part of the uveal tract, which is composed of the iris and ciliary body anteriorly and choroid posteriorly. To list the unique proteins of choroid–RPE, we compared the proteome data obtained in this study with ciliary body proteome data published by our group (Goel et al., 2013) and iris proteome (Murthy et al., 2016). A total of 1147 proteins are found to be unique to the choroid–RPE complex. Some of the unique proteins are rhodopsin kinase (GRK1), S-arrestin (SAG), microphthalmia-associated transcription factor isoform 6, peripherin-2 (PRPH2), LIM homeobox transcription factor 1, beta, and serine/threonine-protein kinase B-raf (BRaf). The representative MS/MS spectra for PRPH2 and GRK1 uniquely identified proteins are given in Supplementary Figure S1.
Peripherin-2
It is a member of the transmembrane 4 superfamily, also known as the tetraspanin family of integral membrane protein. It is a membrane glycoprotein essential for the photoreceptor outer segment disk morphogenesis and maintenance. Defects in this gene are associated with both central and peripheral retinal degenerations (Eriksson et al., 2009; Ferrari et al., 2011).
Rhodopsin kinase
It belongs to G protein-coupled receptor kinase subfamily of the Ser/Thr protein kinase family and initiates desensitization of the receptors by phosphorylation (Bruel et al., 2000). Its kinase activity has been shown to be specifically directed to photoactivated rhodopsin, and it phosphorylates serine residues at positions 334 and 338 (Ohguro et al., 1995). It has been reported to play a role in eye diseases, including Oguchi disease, melanoma, and retinitis pigmentosa (Hayashi et al., 2007).
Identification of “missing proteins”
Finding protein evidence for protein coding genes is a primary mission of the Phase I Chromosome-Centric Human Proteome Project (C-HPP) (Gaudet et al., 2013). We compared our data with the missing protein list provided by C-HPP study (neXtProt release April 28, 2015, application release: 3.0.26). However, in our study, we provided peptide evidence for two such proteins, which were listed as “missing proteins”—vesicular acetylcholine transporter (SLC18A3) and D-dopachrome decarboxylase-like protein (DDTL).
Functional analysis
The biological role of choroid–RPE is transport of ions, metabolism, cell growth and maintenance, thermoregulation, and secretion of growth factors. Functional analysis, against biological processes annotated in HPRD (Muthusamy et al., 2013; Prasad et al., 2009), found the proteins from choroid–RPE to fit into these biological processes. Out of the 5309 proteins identified, it is observed that 18% of proteins are involved in catalytic activity, 6% in transporter activity, 4% in nucleotide and protein binding, 3% in ubiquitin-specific protease activity, 3% in GTPase activity, and 3% in transcription regulatory activity, and rest of the proteins are distributed among the voltage-gated ion channel activity, transmembrane receptor protein tyrosine kinase activity, receptor activity, motor activity, and G-protein coupled receptor activity.
The biological processes for 18% of the proteins remain unspecified. Subcellular localization for the choroid–RPE proteins was annotated as cytoplasm (29%), nucleus (16%), plasma membrane (15%), mitochondrion (9%), extracellular (8%), endoplasmic reticulum (5%), and others (17%). The protein identifications along with their molecular functions and subcellular localization are provided in Figure 3.
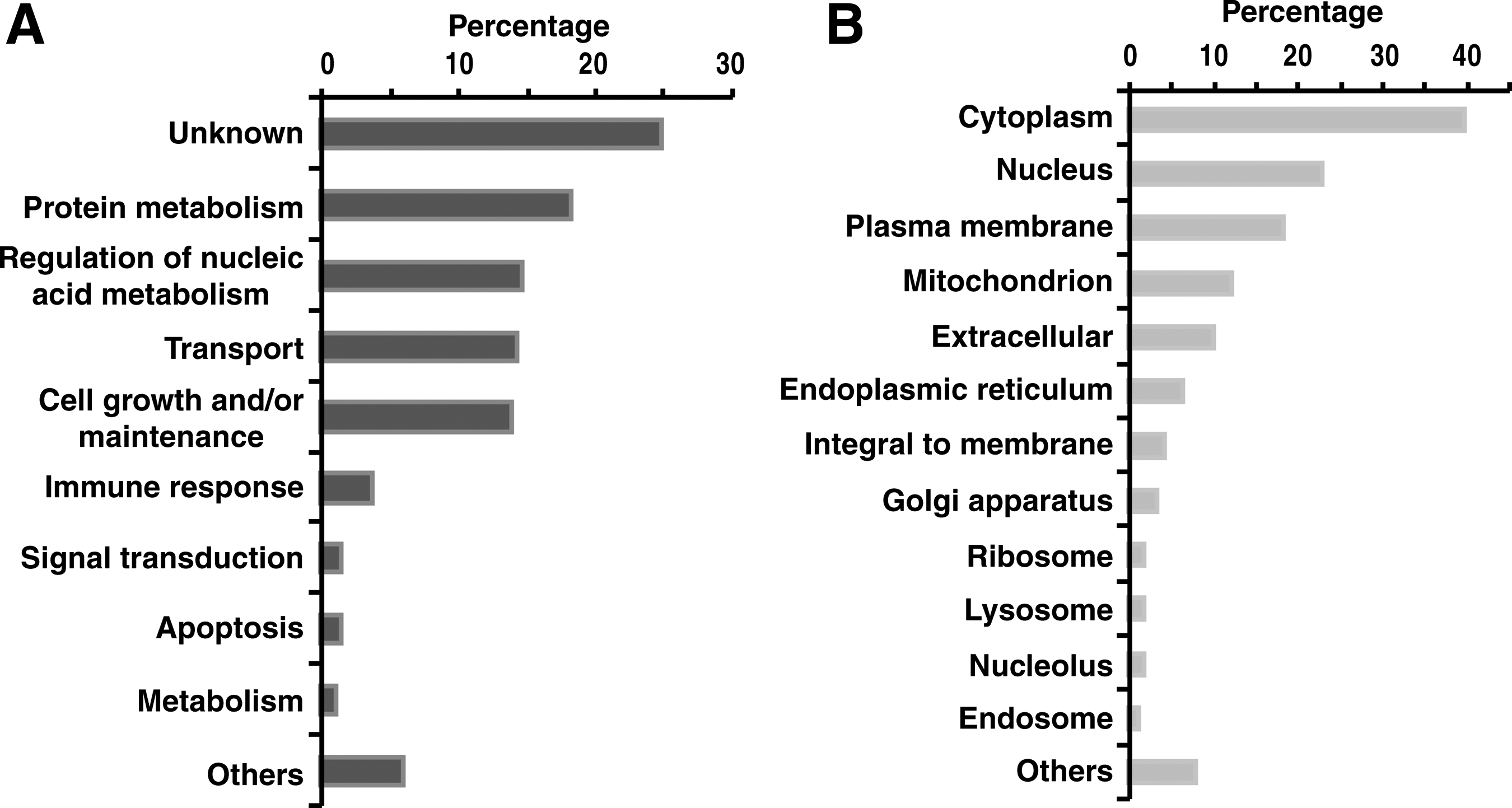
Gene ontology analysis of proteins identified. Molecular functions and subcellular localization.
Canonical pathway and network analysis of the choroid–RPE complex
Protein expression data from choroid–RPE were subjected to IPA to identify potential signaling and metabolic pathways enriched in this tissue. The analysis suggested a significant enrichment of pathways such as EIF2 signaling, integrin signaling, mitochondrial dysfunction, regulation of eIF4 and p70S6K signaling, and clathrin-mediated endocytosis signaling. One of the top canonical pathways in which these proteins participated was mitochondrial dysfunction. One hundred eleven proteins were found to be involved in this pathway (Fig. 4). Mitochondrial dysfunction is the major reason for age-related retinal disease, including macular degeneration and glaucoma (Lee et al., 2011). Eliciting the function of mitochondria in glaucoma pathogenesis may reveal novel therapeutic targets for protecting the optic nerve and preventing vision loss in glaucoma (Kong et al., 2009; Osborne et al., 2014).

Molecules involved in mitochondrial dysfunction. Proteins identified in this study are highlighted.
Protein abundances in the choroid proteome
The peak intensity-based semiquantification method iBAQ (Schwanhausser et al., 2011) was applied to estimate the abundances of proteins in the choroid–RPE complex (Kelkar et al., 2014). A total of 5238 proteins were quantified in the human choroid tissue by iBAQ. The top most abundant proteins in the choroid–RPE were hemoglobin, actin, albumin, histone, vimentin, peroxiredoxin, and tubulin, accounting for 60% of the total choroid protein content. About 250 most abundant choroid proteins constituted 90% of the total protein content. The relative abundance of identified proteins is shown in Figure 5.
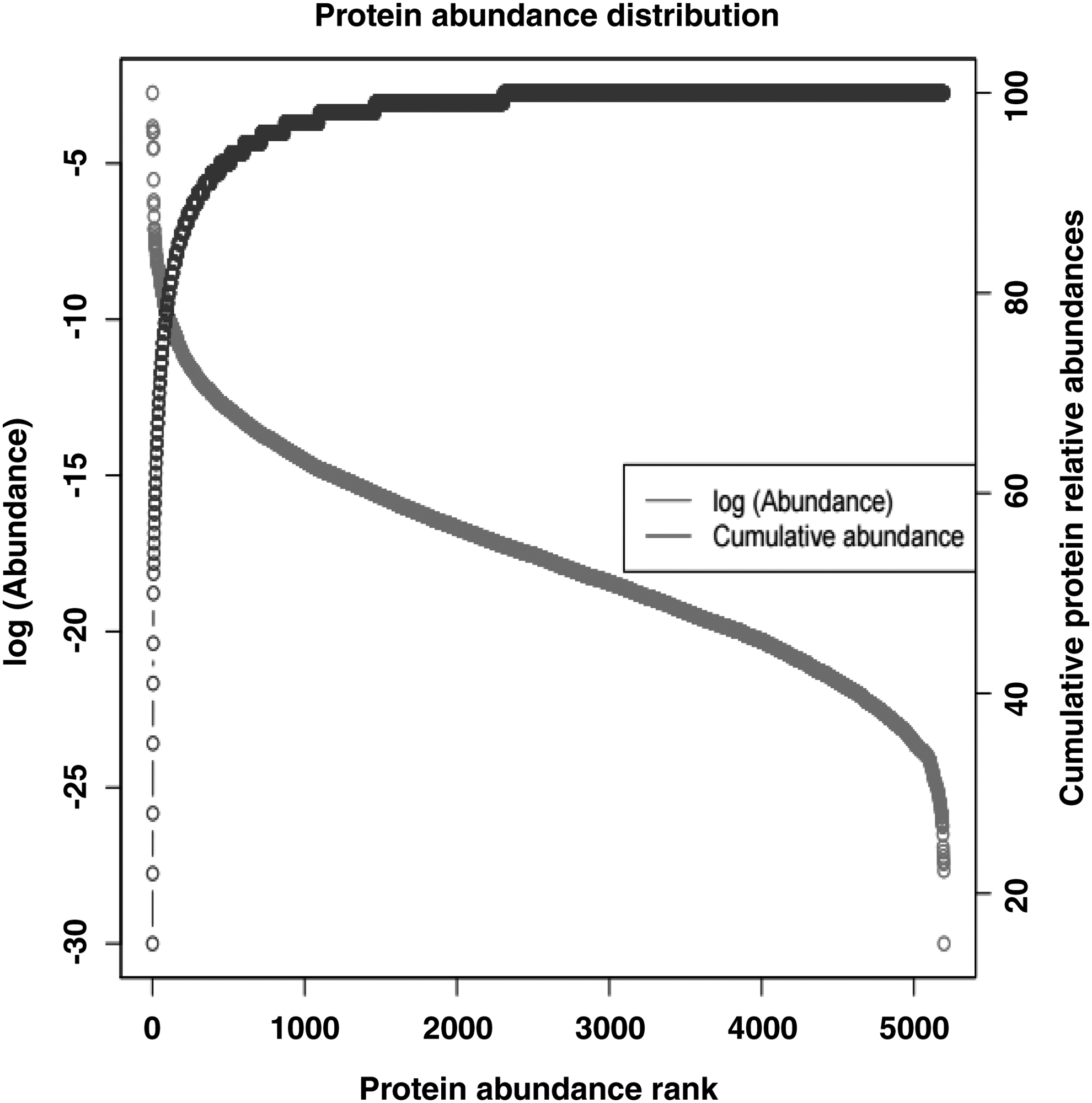
Protein abundances in the choroid proteome. A total of 5238 proteins were quantified by iBAQ and sorted by abundance. The transverse axis shows the protein abundance rank, with a lower rank number indicating higher protein abundance. The left vertical axis shows the protein abundances (red line). The right vertical axis presents the cumulative relative protein abundances along the protein abundance ranks, which indicates that a few high-abundance choroid proteins constitute most of the total protein content (blue line). iBAQ, intensity-based absolute quantification.
Conclusions and Future Outlook
This study provides an in-depth proteomic analysis of the choroid–RPE tissue. These data may serve as an additional resource to the scientific community for better understanding of proteome of human choroid–RPE tissue and help as a baseline for the discovery of potential biomarkers in the diagnosis of uveal disorders. Future research is required to understand the role of these proteins in the choroid–RPE complex. The data from this study are a significant supplement to the Human Eye Proteome Project, which aims at characterizing the proteome of the human eye in health and disease to understand the pathophysiology of eye diseases.
Footnotes
Acknowledgments
All authors have met the ICMJE criteria for authorship. We thank the Department of Biotechnology (DBT), Government of India, and the Infosys Foundation for research support to the Institute of Bioinformatics; M.D. is on deputation from Siddaganga Institute of Technology, Tumkur, to pursue his research work at IOB toward PhD degree under the Faculty Improvement Program of Sree Siddaganga Education Society, Tumkur; R.S.N. is a recipient of Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India; G.D. is a recipient of Senior Research Fellowships from University Grants Commission (UGC), Government of India.
A.K.M. is the recipient of BINC Senior Research Fellowship from Department of Biotechnology (DBT), India. H.G. is a Wellcome Trust/DBT India Alliance Early Career Fellow. Dr. T.S. Keshava Prasad and Dr. Krishna R. Murthy are the recipients of a research grant on “Proteomics based identification and validation of urinary Biomarkers for Age-related Macular Degeneration” from DBT. Dr. T.S. Keshava Prasad is a recipient of a research grant on “Development of Infrastructure and a Computational Framework for Analysis of Proteomic Data (BT/01/COE/08/05)” from DBT.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.