
Abstract
Abstract
Hearing impairment (HI) is one of the leading causes of disability in the world, impacting the social, economic, and psychological well-being of the affected individual. This is particularly true in sub-Saharan Africa, which carries one of the highest burdens of this condition. Despite this, there are limited data on the most prevalent genes or mutations that cause HI among sub-Saharan Africans. Next-generation technologies, such as targeted genomic enrichment and massively parallel sequencing, offer new promise in this context. This study reports, for the first time to the best of our knowledge, on the prevalence of novel mutations identified through a platform of 116 HI genes (OtoSCOPE®), among 82 African probands with HI. Only variants OTOF NM_194248.2:c.766-2A>G and MYO7A NM_000260.3:c.1996C>T, p.Arg666Stop were found in 3 (3.7%) and 5 (6.1%) patients, respectively. In addition and uniquely, the analysis of protein–protein interactions (PPI), through interrogation of gene subnetworks, using a custom script and two databases (Enrichr and PANTHER), and an algorithm in the igraph package of R, identified the enrichment of sensory perception and mechanical stimulus biological processes, and the most significant molecular functions of these variants pertained to binding or structural activity. Furthermore, 10 genes (MYO7A, MYO6, KCTD3, NUMA1, MYH9, KCNQ1, UBC, DIAPH1, PSMC2, and RDX) were identified as significant hubs within the subnetworks. Results reveal that the novel variants identified among familial cases of HI in Cameroon are not common, and PPI analysis has highlighted the role of 10 genes, potentially important in understanding HI genomics among Africans.
Background
H
Genetic factors are estimated to account for 50% of variability in the congenital HI phenotype with a mostly nonsyndromic and autosomal recessive (AR) mode of inheritance (Hutchin et al., 2005). To date, 90 genes have been implicated in nonsyndromic hearing impairment (NSHI) (http://hereditaryhearingloss.org/main.aspx?c=.HHH&n = 86163, accessed December 1, 2016). In many populations, pathogenic variants in GJB2 (connexin 26 gene) are a major contributor to autosomal recessive NSHI (ARNSHI) (Chan and Chang, 2014). However, the prevalence of GJB2- or GJB6-related NSHI is approximating to zero in most sub-Saharan African populations and little is known about the contribution to HI by other known NSHI genes (Bosch et al., 2014a, 2014b; Gasmelseed et al., 2004; Javidnia et al., 2014; Kabahuma et al., 2011; Lasisi et al., 2014).
Targeted genomic enrichment (TGE) and massively parallel sequencing (MPS), for example, with OtoSCOPE®, include 116 HI genes, and has been shown to be an efficient tool for genetic testing for NSHI (Shearer et al., 2010). Using OtoSCOPE, genetic testing of ten multiplex Cameroonian families segregating NSHI identified pathogenic variants in 7 out of 10 families (70%) (Lebeko et al., 2016); within the 7 families, 12 pathogenic variants in 6 genes previously implicated in NSHI were identified, of which 5 were novel (41.6%). The report of five novel pathogenic variants highlights the importance of ethnic-specific filtering (Sloan-Heggen et al., 2016) and underscores the need to further investigate these variants among nonfamilial Cameroonian cases of NSHI.
This study investigated the prevalence of these newly identified mutations in a cohort of HI patients from Cameroon and South Africa. In addition, to explore the interaction of various HI genes that could potentially play a role in clinical penetrance and variable expression of the phenotype, as well the epidemiology of HI in the general population, we investigated the relationships between variants identified by OtoSCOPE using bioinformatics analysis of a human protein–protein interaction (PPI) network.
Materials and Methods
Ethics approval and consent to participate
This study was approved by the Human Research Ethics Committee of the University of Cape Town (ethics approval HREC REF: 455/2014) and the Cameroon National Ethics Committee (ethics approval N°123/CNE/SE/2010). Written informed consent was obtained from participants 18 years or older and from parents/guardians of minors with verbal assent from the children.
Patients and inclusion criteria
Patients were recruited from (1) schools of the deaf and outpatient clinics in Cameroon, as previously reported (Wonkam et al., 2013), and (2) from South Africa at the Effata School of the Deaf in Umtata Eastern Cape and were self-identified as Black Xhosa. As previously described, we included, in this study, patients of Black African descent with NSHI of either (1) putative genetic origin, as revealed by one or more affected family members or consanguinity, or (2) unknown origin. We excluded patients with syndromic HI and those with obvious environmental causes such as meningitis, rubella, mumps, measles, severe prematurity and/or birth weight less than 1500 g, neonatal hyperbilirubinemia, neonatal asphyxia, ototoxicity, or severe head trauma (Bosch et al., 2014a).
Clinical evaluation included a comprehensive questionnaire (exposure to noise, ototoxic agents, and familial history) and the diagnosis of sensorineural HI according to current clinical standards, as previously reported (Wonkam et al., 2013). All probands and affected individuals were examined for syndromic features by a medical geneticist and an ophthalmologist. All probands were negative for pathogenic variants in GJB2, GJB6, and GJA1. The total number of patients included in this study was 25 Black Xhosa South Africans and 57 Cameroonians.
Control participants
A total of 250 ethnically matched Cameroonian controls without HI and negative for a family history were recruited, from apparently healthy blood donors in Yaoundé.
Molecular analysis
DNA preparation and polymerase chain reaction amplification
DNA was extracted from whole blood as previously described (Bosch et al., 2014a, 2014b; Wonkam et al., 2013). Polymerase chain reaction (PCR) primers were designed to amplify between 150 and 400 bps encompassing each mutation. Patients' samples were genotyped through singleplex snapshot PCR using a SNaPShot Multiplex Ready Reaction Mix (Applied Biosystems, Foster City, CA, USA). Fragments were resolved on an ABI 3130xl Genetic Analyzer (Applied Biosystems) with the data being analyzed on GeneMapper, Bioedit Sequence Alignment Editor v 7.2.5 23, and Finch TV v 1.4.0 (Washington, DC, USA). Results of genotypes were confirmed by direct Sanger sequencing using a BigDye Terminator v 3.1 Cycle Sequencing Kit (Applied Biosystems) (Fig. 1), at the Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa.

Pedigrees of selected families (4, 6 and 7) and sequence data showing variants that are causal for hearing impairment. Detected variants include the following: homozygous OTOF NM_194248.2:c.766-2A>G
Sequencing analysis and identification of causative mutations
From the previously reported study using OtoSCOPE, the average coverage for the 350,160 targeted base pairs was 99.60% at a greater than 10X depth of coverage. Sequencing reads were aligned to the reference Human genome (Hg19) using the Burrows-Wheeler Aligner (BWA) algorithm as reported (Shearer et al., 2010). The Exome Aggregation Consortium database was used to remove high-frequency variants (i.e., minor allele frequency ≥0.1%), which were unlikely to be pathogenic and obtain variant frequencies in Europeans, Asians, and Africans (Lebeko et al., 2016). The conservation and deleteriousness of the variants were evaluated using the following tools: likelihood ratio test (LRT), mutation assessor, mutation taster, PolyPhen2, PROVEAN, and sorting intolerant from tolerant (SIFT) (Wang et al., 2010).
Molecular modeling
For the five novel variants identified in Cameroonian families, molecular modeling was performed using SWISS-MODEL (Biasini et al., 2014) and Phyre2 (Kelley et al., 2015) (Fig. 2; Supplementary Fig. S1). Specific templates that were used for each protein or domain within which the novel variant lies were previously reported (Lebeko et al., 2016).
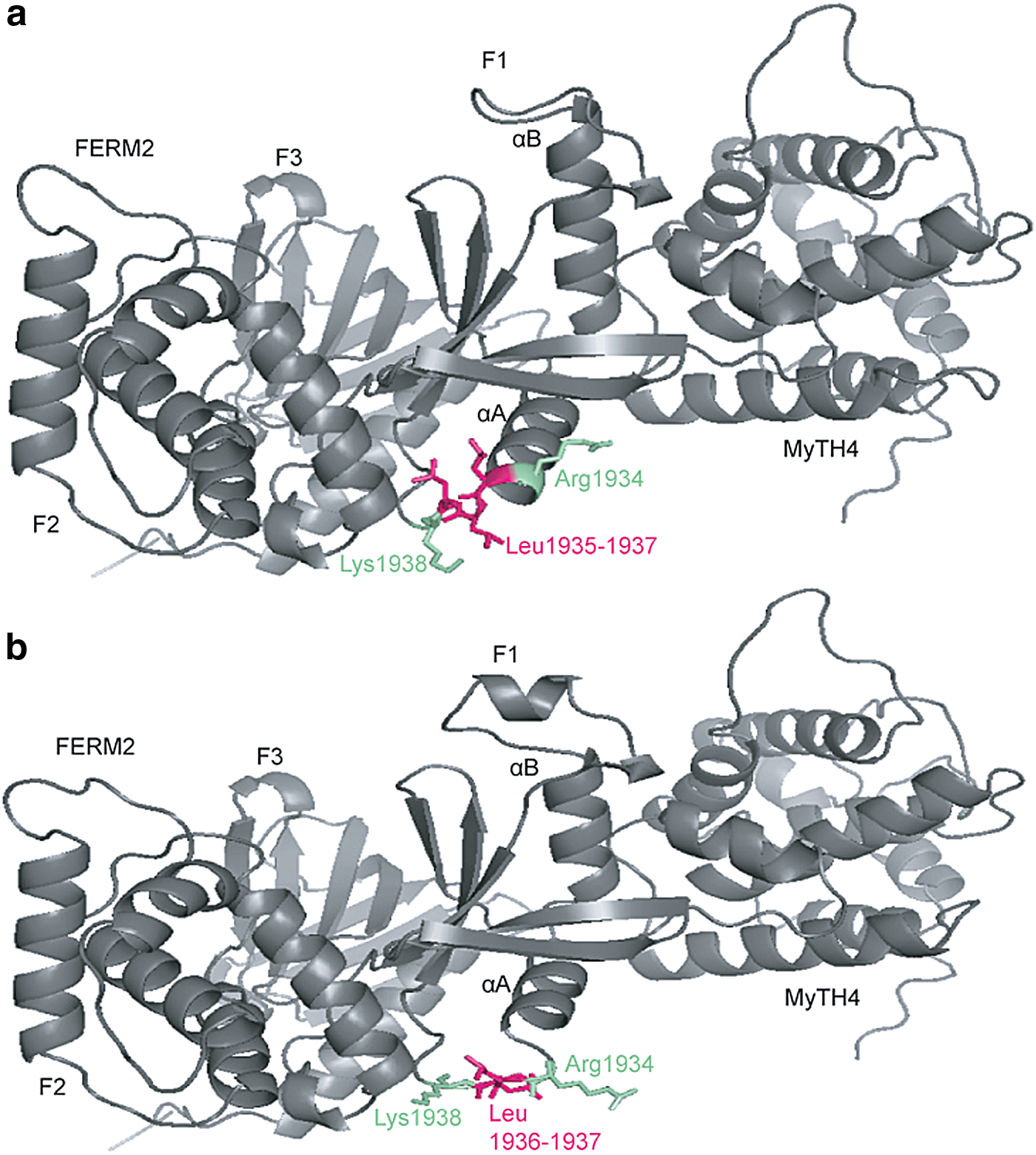
Molecular modeling of MYO7A p.Leu1935del comparing wild-type
Network analysis
Network and enrichment analysis
We used a comprehensive human PPI network (Chimusa et al., 2016; Wu et al., 2009) to understand how the set of casual variants are layered in a biological network. We performed enrichment analysis to examine how these variants and genes within the identified subnetwork are associated with human phenotypes and determine their potential biological processes, pathways, and molecular functions. For this, we used a custom script and two databases: Enrichr (www.amppharm.mssm.edu/Enrichr) and PANTHER (http://pantherdb.org).
To identify potential gene hubs in the network of HI, we queried our reported genes (Lebeko et al., 2016) alongside all other known genes associated with HI in the PPI network; these are the genes which were screened through the OtoSCOPE panel (116 in total). We applied a clustering algorithm in the igraph package of R (www.r-project.org) to determine community structure from the obtained subnetwork of hearing loss interactive genes.
Prioritizing variants on the merged data from all family members
We additionally prioritized variants using the aggregated data of all interrogated family members. We merged all single variants calling files (vcf) from each family member and annotated the resulting merged vcf using ANNOVAR (Wang et al., 2010). Based on prediction approaches implemented in ANNOVAR, we filtered and retained only variants (Supplementary Table S1) that have a functional prediction using 10 approaches (SIFT, PolyPhen 2, MutationTaster, MutationAssessor, LRT, FATHMM, MetaSVM, MetaLR, GERP++, and PhyloP). Functional prediction status included “Deleterious/probably or damaging/disease_causing” (D) or “disease_causing_automatic” (A). Enrichment analysis was performed using two databases, including Enrichr (www.amppharm.mssm.edu/Enrichr) and PANTHER (http://pantherdb.org). We estimated and aggregated the minor allele frequencies (MAF) of single nucleotide polymorphisms (SNPs) downstream and upstream (0 kb, respectively) within each of these retained genes using exome data from 105 Cameroonian control population, all African, European, and East Asian populations from the 1000 Genome Phase 3 Project.
Results
Patients' description
The patients' descriptions are summarized in Table 1. The total number of samples analyzed were from 82 patients with a skew toward male participants (58.5%, n = 48). The majority of affected individuals had nonsyndromic, prelingual, bilaterally profound sensorineural HI, with hearing thresholds between 81 and 119 dB.
N/A, no data available.
Prevalence of previously identified pathogenic variants
Mutations identified through OtoSCOPE in our previous study were not prevalent in this group of patients with NSHI (Table 2). Among Cameroonian individuals with HI, only OTOF NM_194248.2:c.766-2A>G was found in 3 (3.7%) patients with an MAF = 0.02, and MYO7A NM_000260.3:c.1996C>T, p.Arg666Stop was found in 5 (6.1%) patients with an MAF = 0.04 (Table 2). All these variants were in a heterozygous state in all the cases and are thus unlikely to explain, alone, the cause of HI in these patients. None of these variants were found among South African patients. None of the variants previously reported in MYO7A, CDH23, LOXHD1, and SLC26A4 were identified in this group of patients with HI (Table 2).
RefSeq#: MYO7A, NM_000260.3; SLC26A4, NM_000441.1; OTOF, NM_194248.2; LOXHD1, NM_144612.6; CDH23, NM_022124.5.
cDNA, complementary DNA.
Molecular modeling
Previous molecular modeling data reported changes to the LOXHD1 protein that affected binding (Lebeko et al., 2016). The effect of the novel MYO7A variant results in a disruption of the folding of the FERM2 domain needed for binding to the SANS protein central domain disrupting their dimerization (Fig. 2) (Wu et al., 2011). The p.Asp2133Glu change in CDH23 results in a loss of calcium binding site which is integral to the functioning of the protein (Supplementary Fig. S1b) (Sotomayor et al., 2010). Coupled with the change in linker regions caused by the p.Met2907Thr substitution (Supplementary Fig. S1d) (Patel et al., 2006), the protein is rendered nonfunctional.
Gene network analysis and enrichment of reported genes
Querying the genes reported in Table 2 in the human PPI network, we identified 16 pairs of interactive genes (Fig. 3A). Interestingly, the novel mutation identified in MYO7A appears to be the central hub of the subnetwork, indicating a major functional role that the variant may play in HI. The subnetwork (Fig. 3A) was not found to be significantly associated with any biological pathway; however, from the OMIM (Online Mendelian Inheritance of Man database) disease database (www.omim.org), the whole subnetwork is enriched with Usher Syndrome and deafness variants (p = 4.134e-7 and 0.0001138, respectively). Two biological processes were found to be significantly associated with the subnetwork, including sensory perception of mechanical stimulus (GO: 0050954, p = 1.430e-8) and sensory perception of sound (GO: 0007605, p = 1.246e-8). Furthermore, 60% of the molecular functions (Fig. 3B) of these variants are in binding (GO: 0005488) and/or structural molecule activity (GO: 0005198) pathways.
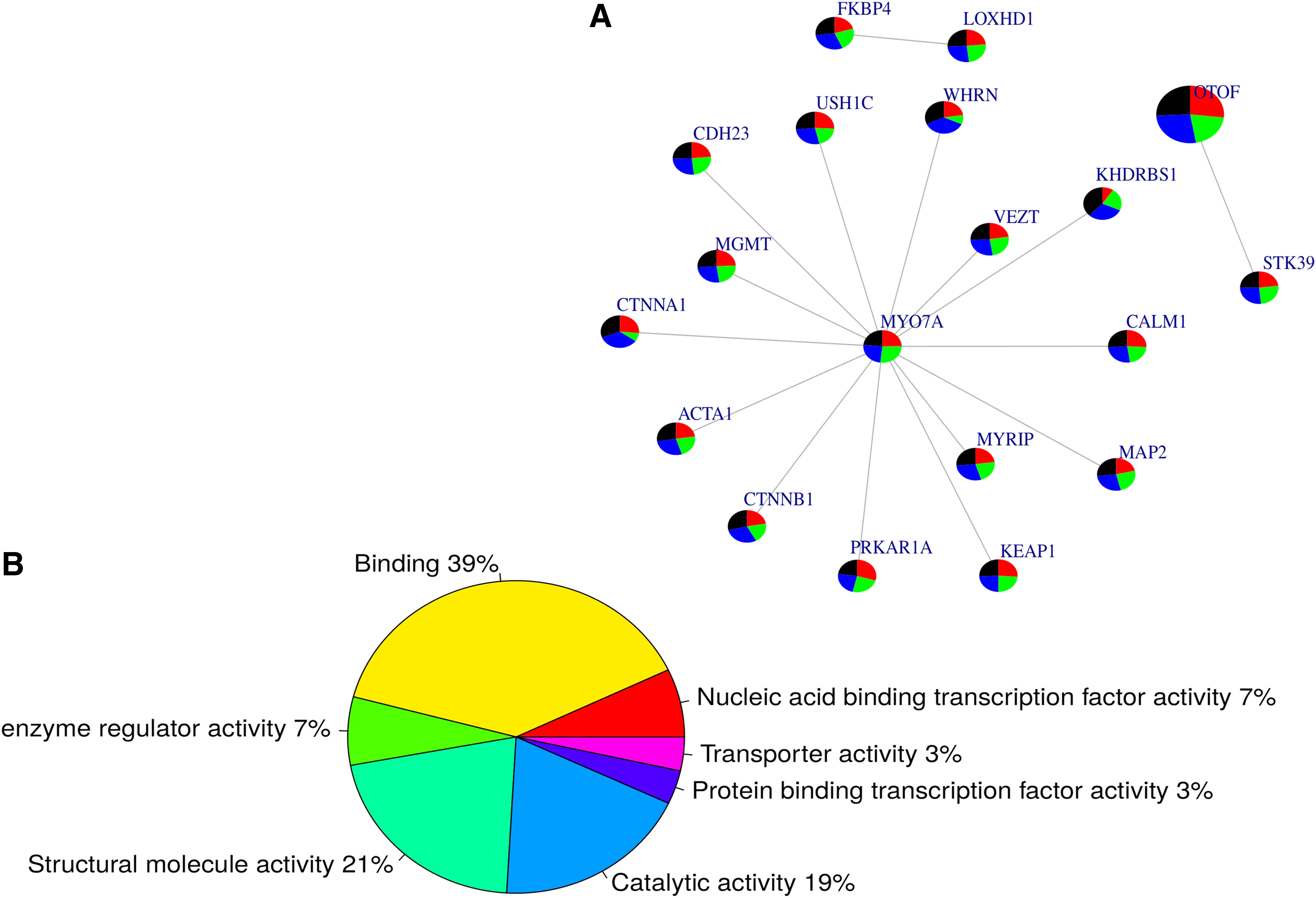
Subnetwork of interactive genes with potential mutations associated with hearing loss. From the identified causal mutations occurring in genes displayed in Table 2 and Supplementary Table S1, we used a human protein–protein interaction network to interrogate how these genes are positioned in a biological network: we identified 16 pairs of interacting genes
From the compiled gene network of 116 hearing loss genes, we identified 10 hubs (the most import gene node, harboring the major connectivity within the subnetwork), including MYO7A, MYO6, KCTD3, NUMA1, MYH9, KCNQ1, UBC, DIAPH1, PSMC2, and RDX (Fig. 4A). The subnetwork (Fig. 4B) is significantly associated with known biological pathways such as proteasome degradation, Tumor necrosis factor (TNF)-alpha signaling pathway, and transforming growth factor (TGF)-beta signaling pathway (Fig. 4B). Sixty-three percent of the genes within the subnetwork (Table 3) have a molecular function in binding (GO: 0005488) and/or catalytic activity (GO: 0003824) pathways, and are significantly enriched with deafness (p = 4.22e-15).
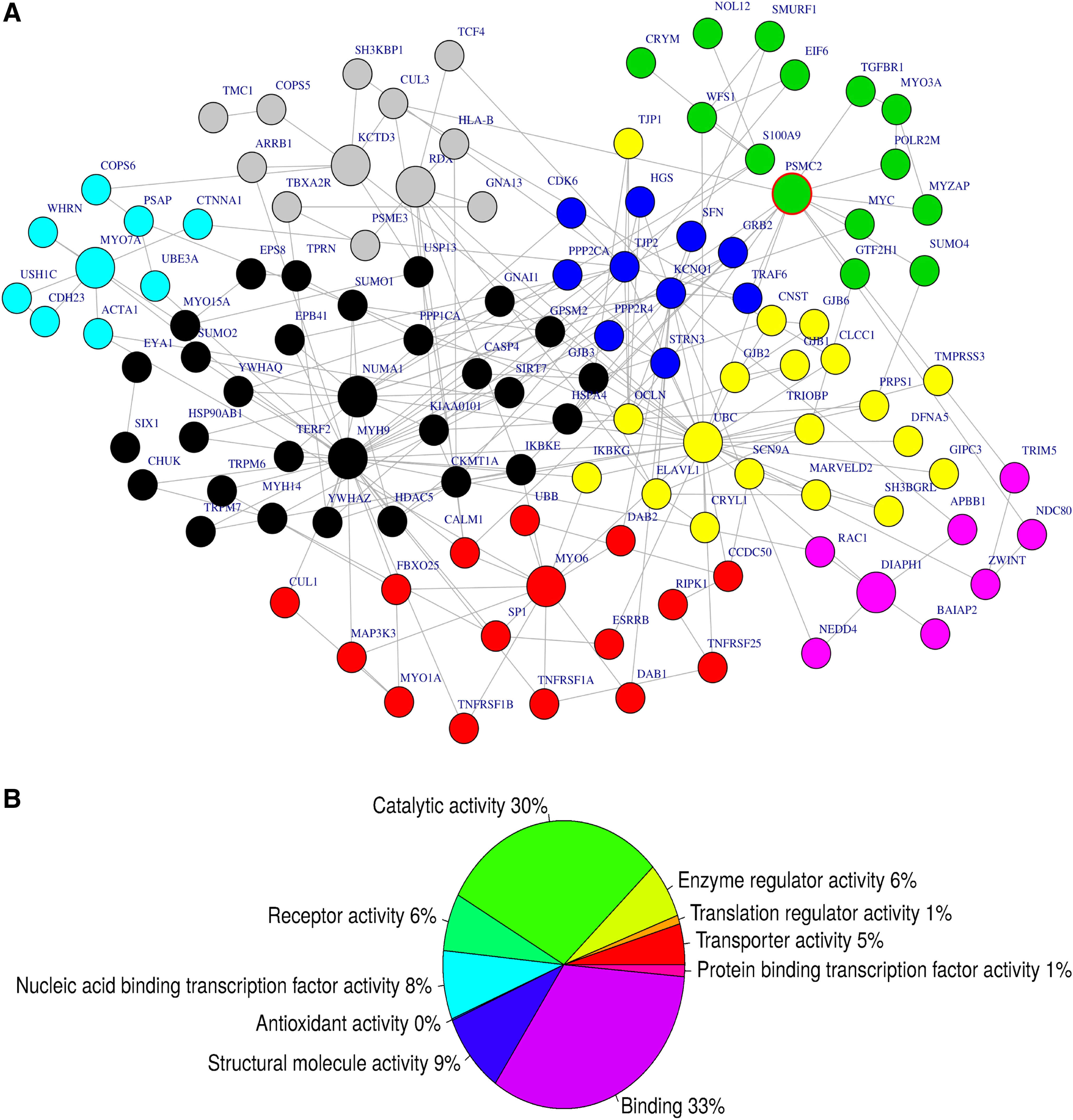
Subnetwork of interacting genes with mutations potentially associated with hearing loss. We identified 10 hubs from the subnetworks extracted from protein–protein interaction database
Pathway enrichment using Enrichr (www.amppharm.mssm.edu/Enrichr), based on genes in the subnetwork in Figure 4A; 63% of the genes within the subnetwork are annotated to the molecular functions binding (GO: 0005488) and/or catalytic activity (GO: 0003824) and are significantly enriched with deafness (p = 4.22e-15).
TNF, tumor necrosis factor; TGF, transforming growth factor.
Leveraging the combined data in prioritizing variants and gene enrichment
We merged the data of nine Cameroonian family members, and filtered and prioritized variants using the approach and criteria described above. We replicated OtoSCOPE findings in two genes, OTOF and LOXHD1 (Supplementary Fig. S2 and Supplementary Table S1), and identified other genes in linkage disequilibrium (LD) with those reported in Table 2. Furthermore, we used a comprehensive human PPI network (Chimusa et al., 2016; Wu et al., 2009) to derive their biological network positions. We identified 29 pairs of interacting genes (Supplementary Fig. S2A). From the OMIM disease database (www.omim.org), the whole subnetwork is enriched with HI variants (p = 0.000001870 and adjusted p = 0.000013).
The highest proportion of variants in the subnetwork (Supplementary Fig. S2A) is associated with endoplasmic reticulum pathways (Supplementary Fig. S2B) and 60% of the molecular functions (Supplementary Fig. S2C) of these variants are related to binding (GO: 0005488) and/or catalytic activity (GO: 0003824). Using exome data of 105 Cameroonian controls, as well as data extracted from the 1000 Genomes project, results displayed in Supplementary Table S1 indicate that the variants in the majority of these hub genes displayed almost similar MAFs across various populations, with the exception of variants in MYO1A, which were significantly more prevalent among East Asians, and variants in GIPC3, which were relatively rare among the East Asian populations.
Discussion
HI is one of the highest contributors to disability worldwide, impacting on the social, economic, and psychological well-being of the affected individual (Olusanya, 2011). This is particularly true in low-income regions such as sub-Saharan Africa, which also carries one of the highest burdens of this disease (WHO, 2012). Despite this, there are limited data to support the most prevalent genes or mutations that cause HI among patients from this region. Using TGE and MPS in 10 Cameroonian families recently demonstrated the importance of this approach in the resolution of genetic aetiology of HI in Africa and it role in uncovering novel variants (Lebeko et al., 2016). Follow-up research among nonfamilial cases of HI, potentially of genetic origin as presented in this study, revealed that the aforementioned variants are not common in other nonfamilial cases of HI in Cameroon, and absent from a small sample of Xhosa patients from South Africa.
The findings of the heterozygous state of OTOF NM_194248.2:c.766-2A>G found in three patients, and MYO7A NM_000260.3:c.1996C>T, p.Arg666Stop found in 5 (6.1%) patients (Table 2) suggest that these eight individuals are most likely compound heterozygotes, with second mutations to be investigated in these genes. The relatively high number of NSHI genes and novel variants among a relatively small sample of Cameroonians (Lebeko et al., 2016) demonstrates the high level of genetic heterogeneity, and could reflect genetic diversity found in Cameroons, which mimics many populations in sub-Saharan Africa (Lambert and Tishkoff, 2009).
The absence of a mutation in 30% of Cameroonian families previously studied indicates the possibility of discovering new HI-associated genes in Africans (Lebeko et al., 2016). Supporting this, recent studies using MPS that looked at the exons of 180 genes in various populations across the globe, including two sub-Saharan African populations, showed the lowest number of resolved cases, reported at 4% and 17%, among Nigerian and South African populations, respectively (Yan et al., 2016). A similar trend with only 26% resolution rate was reported among African-Americans, the lowest when compared to that of other ethnic groups (Sloan-Heggen et al., 2016). Thus, in an effort to address the genetic causes of HI among patients of African descent, the use of whole-exome sequencing should be the next approach. Estimates indicate that at least 1000 NSHI genes remain to be identified (OMIM) (Hertzano and Elkon, 2012), and African populations could be important in this endeavor.
The MYO7A variant c.5880_5882 delCTT (p.Phe1963del) was previously identified in a French patient with Usher syndrome (Roux et al., 2006), and bioinformatics analysis performed in this study also revealed that the subnetwork associated with this MYO7A variant is enriched with Usher syndrome annotation (p = 4.134e-7). While surveillance will determine whether retinitis pigmentosa will develop in the Cameroonian hearing-impaired probands and affected siblings, it is possible that the compound heterozygous variants in MYO7A are causal of NSHI. Two previously reported MYO7A tail variants caused NSHI rather than Usher syndrome (Riazuddin et al., 2008), like the novel MYO7A p.Leu1935del variant reported in this study.
Likewise, for CDH23 compound heterozygous variants (Table 2), the occurrence of an NSHI variant in trans with an Usher allele is predicted to cause NSHI only (Schultz et al., 2011). Similarly, the compound heterozygous SLC26A4 variants identified in one of the Cameroonian families (family 6) (Fig. 1; Table 2) were previously identified in Chinese NSHI probands (Jiang et al., 2015; Yasunaga et al., 2000; Yuan et al., 2012) and are unlikely to cause Pendred syndrome. A homozygous splice mutation, OTOF [c.766-2A>G], identified in this study (family 4; Fig. 1) was previously reported in a southwest Indian family segregating NSHI (Yasunaga et al., 2000).
The bioinformatics interrogation of genes in the human PPI network performed in this study could give us new insight into biological process involved in HI, in understanding variable clinical phenotypes, and hopefully, investigating new routes for therapeutic interventions. As expected, there was a significant association between the reported subnetwork and the biological processes' sensory perception of sound and mechanical stimulus. Specifically, the network analysis identified 10 hubs that involved genes that could happen in future studies to be of importance in Cameroon. In addition, the analysis has reaffirmed the importance of MYO7A in HI. MYO7A is a large gene that spans 87 kb of the genome and plays an important role in nonsyndromic as well as syndromic HI (i.e., Usher syndrome) (Gibbs et al., 2010). OtoSCOPE interrogates the exons of genes known to be associated with HI, thus any variants identified within the genes could potentially have clinical implications (Shearer et al., 2013).
The above PPI analysis results on the variants reported by OtoSCOPE, in addition to analysis of data extracted from the 1000 Genomes with a community network analysis across the different populations, have also shown that for some targeted HI genes, the overall MAFs at the gene level were population-specific common (Supplementary Table S1). Last, prioritization of genes and their variants from merged data replicated OTOF and LOXHD1 as important genes in HI, which also appeared to be occurring in LD with other reported variants. Much like MYO7A, OTOF carries variants that are reported in multiple populations (Table 2) and could prove to be important genes in HI across different populations, and their roles and interactions should be further investigated.
Conclusions and Outlook
The result reveals that novel variants recently identified among selected familial case of HI in Cameroon variants are not common in isolated cases of NSHI. PPI network analysis has highlighted the potential role of 10 genes in HI genomics and phenotypes among Cameroonians that need further investigation. It is anticipated that future ramifications of the study will include study of exomes data of families segregating HI, isolated probands with HI, as well as control populations without HI from Africa, with the anticipation that the genes highlighted by the present studies could play prominent roles in the etiology and epidemiology of HI in sub-Saharan Africa. Such knowledge will assist in establishing appropriate molecular diagnosis from this condition, as well as clinical and molecular clues to be used for anticipatory guidance in clinical settings, based on genotypes to phenotypes correlation studies.
Footnotes
Acknowledgments
The authors thank parents and patients who have participated in this research and the following organizations for their help and support with data collection: Faculty of Medicine and Biomedical Sciences, University of Yaoundé 1; and Faculty of Health Sciences, University of Cape Town. The research was funded by the University of Yaoundé for clinical phenotyping and DNA extraction and the National Health Laboratory Services Research Grant, South Africa, and the Medical Research Counsel Self-Initiated Research Grant, South Africa. Kamogelo Lebeko was supported by funding provided by the University of Cape Town and the National Research Foundation, South Africa. The dataset supporting the conclusions of this article, including raw sequencing and clinical data, is available from authors upon request.
A.W. conceived the study, participated in its design, interacted with the patients, coordinated the blood sample collection, and drafted the article. K.L., M.N., E.C., N.M., C.D., and A.W. carried out the genetic studies, performed the data analysis, and contributed to drafting the article. K.L. and C.D. participated in its design, interacted with the patients, coordinated the blood sample collection, and helped to draft the article. E.C., N.M., and A.W. performed the bioinformatics analysis, coordinated the data interpretation and statistical analysis, and helped to draft the article. All authors read and approved the final article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.