
Abstract
Abstract
Metabolic systems engineering is being used to redirect microbial metabolism for the overproduction of chemicals of interest with the aim of transforming microbial hosts into cellular factories. In this study, a genome-based metabolic systems engineering approach was designed and performed to improve biopolymer biosynthesis capability of a moderately halophilic bacterium Halomonas smyrnensis AAD6T producing levan, which is a fructose homopolymer with many potential uses in various industries and medicine. For this purpose, the genome-scale metabolic model for AAD6T was used to characterize the metabolic resource allocation, specifically to design metabolic engineering strategies for engineered bacteria with enhanced levan production capability. Simulations were performed in silico to determine optimal gene knockout strategies to develop new strains with enhanced levan production capability. The majority of the gene knockout strategies emphasized the vital role of the fructose uptake mechanism, and pointed out the fructose-specific phosphotransferase system (PTSfru) as the most promising target for further metabolic engineering studies. Therefore, the PTSfru of AAD6T was restructured with insertional mutagenesis and triparental mating techniques to construct a novel, engineered H. smyrnensis strain, BMA14. Fermentation experiments were carried out to demonstrate the high efficiency of the mutant strain BMA14 in terms of final levan concentration, sucrose consumption rate, and sucrose conversion efficiency, when compared to the AAD6T. The genome-based metabolic systems engineering approach presented in this study might be considered an efficient framework to redirect microbial metabolism for the overproduction of chemicals of interest, and the novel strain BMA14 might be considered a potential microbial cell factory for further studies aimed to design levan production processes with lower production costs.
Introduction
L
In addition, it is biodegradable and proposed as an ideal raw material for the production of green plastics (Wu et al., 2013). However, levan could never find its proper place in the industrially produced polymer market due to lack of economical large-scale production schemes (Oner et al., 2016).
A variety of microorganisms, including Acetobacter, Azotobacter, Bacillus, Brenneria, Brachybacterium, Erwinia, Geobacillus, Lactobacillus, Pseudomonas, and Zymomonas produce levan as an exopolysaccharide (EPS) from sucrose-based substrates (de Paula et al., 2008; Inthanavong et al., 2013; Jathore et al., 2012; Keith et al., 1991; Liu et al., 2017; Moussa et al., 2017; Revin et al., 2016; Srikanth et al., 2015; Tian et al., 2011; Waldherr et al., 2008). In addition, Halomonas smyrnensis AAD6T has been reported as a levan producer halophilic bacterium (Poli et al., 2013). On the other hand, wild-type strains have limited efficiency in the development of economical production processes due to their relatively low yields and production rates. Therefore, there is an urgent need for novel strain development strategies for levan overproduction.
With the aim of transforming microbial hosts into cellular factories, metabolic systems engineering is being used to redirect microbial metabolism for the overproduction of chemicals of interest. In silico genome-scale modeling of metabolism has proven to be useful in this field (Simeonidis and Price, 2015). Being a useful guide for identification and filling of knowledge gaps, once a metabolic network is reconstructed, mathematical methods such as convex analysis and linear programming can be applied to simulate the cellular behavior under different genetic and physiological conditions, and to develop metabolic engineering strategies for the construction of strains with desired and improved properties.
Metabolic engineering of EPS-producing strains has been successfully performed, for example, for the overproduction of xanthan (Bajaj et al., 2007), gellan (Bae et al., 2004), and cellulose (Becker and Wittmann, 2012a). Moreover, systems metabolic engineering pioneers new possibilities for the constitution of bio-based polymers at the desired titers, yields, and productivities, and is currently available to facilitate design and improvement of the performance of microorganisms for fermentative production of an increasing number of chemicals, materials, and fuels (Becker and Wittmann, 2012b; Jang et al., 2012; Tanimura et al., 2013).
The availability of whole genome sequence information (Diken et al., 2015; Sogutcu et al., 2012) makes H. smyrnensis AAD6T a good candidate as a microbial cell factory for systems metabolic engineering studies toward development of novel strains with improved levan biosynthesis. In this study, the genome-scale metabolic model of AAD6T (Diken et al., 2015) was used to determine possible strain improvement strategies for levan overproduction. In silico simulations proposed the fructose uptake mechanism as an efficient target for further metabolic engineering studies. Therefore, we performed insertional mutagenesis and triparental mating techniques to construct a novel, metabolically engineered H. smyrnensis strain, BMA14, with improved levan production capability.
Materials and Methods
Microorganisms, plasmids, and culture conditions
Halomonas smyrnensis AAD6T is a halophilic bacterium that was isolated from soil samples taken from Camaltı Saltern Area, in Izmir, Turkey (Poli et al., 2009). The wild-type strain was used as a starter host for genetic manipulations. Escherichia coli TOP10 competent cells were used for transformations. Plasmids employed in this study are shown in Table 1. The basal medium for H. smyrnensis AAD6T consists of 137.2 g/L NaCl; 50 g/L sucrose; 7 g/L K2HPO4; 2 g/L KH2PO4; 0.1 g/L MgSO4 · 7H2O; 1 g/L (NH4)2SO4; and 0.5 g/L peptone. Trace element solution, consisting of 0.36 g/L MnCl2 · 4H2O, 0.44 g/L ZnSO4 · 7H2O, 2.3 g/L FeSO4 · 7H2O, and 0.05 g/L CuSO4 · 5H2O, was filter sterilized and added as 0.1% (v/v) to the medium. Filter-sterilized thiamine solution was added at 0.8 mg/L final concentration. SW-2 medium for conjugation was prepared as reported in Ref. Vargas et al., 1997.
HPr, histidine-containing phoshocarrier protein.
Unless otherwise stated, cultures were incubated at 37°C in an orbital shaker at 180 rpm. E. coli cells were grown in Luria-Bertoni (LB) medium. When used, filter-sterilized antibiotics were added at the following final concentrations: ampicillin (Amp), 100 μg/mL for E. coli; kanamycin (Km), 100 μg/mL for E. coli and H. smyrnensis AAD6T; and streptomycin (Str), 50 μg/ml for H. smyrnensis AAD6T. Growth was monitored as the optical density (OD) of the culture at 600 nm for E. coli and at 660 nm for H. smyrnensis AAD6T with a Perkin-Elmer Lambda 35 UV/Vis spectrophotometer.
Genome sequence information and genome annotation
The 16S rRNA sequence of H. smyrnensis AAD6T has been deposited in the NCBI database with the accession no. DQ131909.2 (Poli et al., 2009) and the whole genome sequencing project has been deposited in the DDBJ/EMBL/GenBank (DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank) with accession numbers AJKS02000001 to AJKS02000034 (Sogutcu et al., 2012). The gene prediction and annotation were taken from our previous study (Diken et al., 2015). The multiple sequence alignments were performed using Clustal Omega at EMBL server (Sievers et al., 2011).
Identification of optimal strategies for levan overproduction
The genome-scale metabolic model of AAD6T was reconstructed (Diken et al., 2015) based on the published protocol for the generation of genome-scale metabolic models (Thiele and Palsson, 2010). In this study, we recruited the model to determine possible strain improvement strategies for levan overproduction. For this purpose, an in silico knockout study was performed through OptFlux (Rocha et al., 2010) using simulating annealing (SA), evolutionary algorithm (EA), and strength pareto evolutionary algorithm (SPEA2) as the optimization algorithms for strain optimization.
Simulations were performed with four different objective functions: (1) the biomass-product couple yield (BPCY), which calculates the product of the biomass flux and levan production flux, (2) the product yield with minimum biomass (YIELD), which returns the value of the levan production flux divided by the substrate (sucrose) consumption flux, (3) BPCY-BPCY, which represents simultaneous maximization of growth rate and levan production rate, and (4) YIELD-YIELD, which represents simultaneous maximization of growth rate divided by the substrate consumption flux and levan production flux divided by the substrate consumption flux. In this study, in each simulation, a set of genes (with a maximum number of six) was identified to knock out to maximize the production of a desired compound, that is, levan.
Determination of targeted genomic region
Based on genome annotations, the fructose uptake system in H. smyrnensis AAD6T was investigated through web-based software, Interproscan (EMBL), and Conserved Domain Architecture Tool (National Center for Biotechnology Information: NCBI), to identify the subunit and domains of fructose-specific phosphotransferase system (PTSfru). The histidine-containing phoshocarrier protein (HPr) and fructose-specific IIA component complex was selected as targeted region for metabolic engineering.
Genetic manipulations on target HPr region
Halomonas smyrnensis AAD6T genomic DNA was isolated with the Wizard Genomic DNA Purification Kit (Promega) and plasmid DNA was isolated with the High Pure Plasmid Isolation Kit (Roche). Amplifications were carried out by specifically designed primers (Table 2). Restriction enzyme digestion, modifying enzyme treatments, and ligations were performed as recommended by the manufacturers (Promega, NEB and Thermo Scientific). DNA sequencing was performed by Beijing Genome Institution (China).
Arrows (↓↑) indicate the recognition sequence of the restriction enzymes. HPr, histidine-containing phosphocarrier.
HPr region was amplified from genomic DNA with HPr-F/R primers, cloned with pJET1.2/blunt vector (CloneJET PCR Cloning Kit; Thermo Scientific), and transformed into E. coli TOP10 chemically competent cells. One of the transformants was selected for insertional mutagenesis and named as pJet/HPr4.
pHP45Ω-Km (Prentki and Krisch, 1984) containing the Omega-Km (Ω-Km) cassette was employed for insertional mutagenesis. Ω was amplified from pHP45Ω-Km plasmid DNA by OmegaF/R primers. Vector pJet/HPr4 and insert Ω-amplicon were digested with SgrAI restriction enzyme. HPr region had a recognition site for SgrA1 enzyme at the center of its sequence and Ω cassette was amplified with primers that were specifically designed to have SgrAI-specific recognition sites for both ends. After alkaline phosphatase treatment of vector to prevent recircularization of the strand, ligation was performed, and ligation mixture was transformed into E. coli TOP10 competent cells. Transformants were screened by Km resistance, which came from Ω cassette, colony PCR, and sequencing analysis. Resulting plasmid was named as pΩHOM2.
Generation of knockout mutants
The plasmid pΩHOM2 has both HPr region and Ω cassette (HPr::Ω cassette) with right orientation. To recombine the HPr mutations into the H. smyrnensis AAD6T, 3-kb BglII-BamHI fragments from pΩHOM2 were cloned into the pUK4134 (Skrzypek et al., 1993), which carries the gene conferring StrS and thus can function as a suicide vector for microbial strains with StrR background, to give plasmid pUKHom4, which was mobilized into the H. smyrnensis AAD6T wild-type strain by triparental mating. Mutant strains resulting from a double homologous recombination event were identified as StrR and KmR colonies on SW-2 plates containing 10% sucrose, and then tested on optimum medium plates that have corresponding antibiotics. HPr::Ω cassette was amplified from pΩHOM2 plasmid DNA with specifically designed HOM-F/R primers that have recognition sites for BglII enzyme.
Then, pUK4134 suicide vector was digested with BamHI, whereas insert HPr::Ω cassette was digested with BglII. These two restriction enzymes were selected to make compatible ends between the vector and insert. Ligation mixture was used directly for transformation. Screening by nucleotide sequencing and antibiotic-aided selection was performed for transformants to determine the recombinant that has the correct insert. After all these analyses, one colony, which was named as pUKHom4, was selected for conjugation.
Conjugal transfer of plasmids by triparental mating
Plasmid transfers were performed by triparental mating. The recipient strain, H. smyrnensis AAD6T, was subcultured 1:50 the day before conjugation and grown. Helper (pRK2013) (Figurski and Helinski, 1979) and donor strains (pUKHom4) were subcultured 1:10 for 2 h before conjugation to obtain early stationary-phase cultures. The recipients were centrifuged and washed thrice in SW-2 (similar to SW-10 medium, but containing 2% total salts) medium to eliminate any remains of antibiotics as described by Vargas et al. (1997). Helper and donor strains were centrifuged and suspended in an equal volume of the same medium used for the recipient. Resuspended helper, donor, and recipient cells were mixed 1:1:1 by volume and the mixture was spotted on a nitrocellulose filter membrane (pore size 0.45 μm; Sigma Aldrich) placed on the agar surface of a well-dried SW-2 medium.
The mixture was also placed as drops directly on the surface of the plate. The mixtures were incubated overnight at 37°C. After incubation, the cells were collected in 1 mL of the optimum growth medium containing 13% NaCl and spread on plates with proper concentration of the antibiotic used for selection. After overnight incubation of conjugation, colonies were transferred into antibiotic-aided plates and incubated for 2 days. At the end of 2 days of incubation, resultant colonies were selected and then retransferred again to ensure their resistance with respect to passaging. Transconjugants were screened with antibiotic resistance and selected with both colony PCR and sequencing. PCR amplification was performed with these survivor colonies with omega F/R primers to ensure the insertion of Ω cassette into the target HPr region.
Susceptibility tests
Susceptibility tests were performed using Amp, Km, and Str. Growth on plates was monitored by increasing antibiotic concentrations from 25 to 100 μg/mL. Agar plates were inoculated with 50 μL of H. smyrnensis AAD6T culture at OD660 of 0.8, and then incubated at 37°C for 1–4 days.
Growth profiles
All fermentation media components were dissolved in distilled water, and then sterilized separately by autoclaving. The pH was adjusted to 7.0 using sterile 1 M NaOH or with 1 M HCl. Sterile salt and carbon source solutions were combined to 1 L final volume. Two hundred milliliter of optimum medium was inoculated with H. smyrnensis AAD6T (wild type) and 200 mL of optimum medium with corresponding antibiotics (Km and Str) was inoculated with BMA14. The growth profiles of AAD6T and BMA14 were monitored by measuring the OD660. A cell-free medium was used as the blank. Growth was terminated at the end of 154 h.
Levan production
The basal medium was inoculated with AAD6T and BMA14 precultures separately. Fermentation was terminated after 154 h. Cells were harvested at 10,000 rpm for 20 min. The supernatant phases, containing levan, were treated with two volumes of ethanol, held at −20°C overnight, and then centrifuged at 12,000 rpm and 4°C for 30 min. The pellets were dissolved in warm distilled water. Dissolved polymer was dialyzed against several runs of distilled water for 3–5 days. After dialysis, levan that was produced by BMA14 and AAD6T was dried at 60°C overnight. All the experiments were performed in duplicate.
Sucrose assay
Sucrose concentration analysis was performed by Agilent 1100 high-performance liquid chromatography (HPLC) system with refractive index detector using the Zorbax Carbohydrate Analysis Column 4.6 × 250 mm (Agilent). All samples collected from the fermentation broth were centrifuged at 10,000 rpm for 20 min to precipitate cells. The cell-free samples were filtered through 0.2 μm filter before use, and an equal volume of 50:50 acetonitrile:water solution was added. The flow rate was 1.4 mL/min, injection volume was 3 μL, the mobile phase was 75:25 acetonitrile:water, and the temperature of the column was 30°C.
The theoretical carbon recovery calculation was based on the molecular formula of compounds from the KEGG COMPOUND database and the elemental composition of the bacteria from Sucharitha et al. (2017).
Glucose assay
Extracellular glucose concentrations were determined using the enzymatic kit (Sigma Aldrich). The intensity of the pink color measured at 540 nm was proportional to the original glucose concentration. Distilled water was used as blank. Standards were prepared with glucose solutions at known concentrations, then a calibration curve was plotted and glucose concentrations of samples were calculated by using the equation that was derived from the calibration curve.
Statistical evaluations
All parameters were represented by mean values ± standard errors of the means. Statistical comparisons of mean values were carried out through Student's t-test, and p-value <0.05 was considered significant.
Results
Gene/reaction knockout analysis for metabolic engineering targets
To determine possible optimal phenotypes of the bacteria, in silico knockout simulations were performed within a multiobjective optimization framework by use of the metabolic model of AAD6T, four different objective functions, and three different optimization algorithms. The multiple solutions (i.e., gene knockout strategies) obtained through utilization of different optimization algorithms and objective functions were merged together, and removal of duplicates resulted with a total of 495 knockout strategies up to six gene deletions to improve levan production capability.
Comparative analysis of these gene knockout strategies resulted with the observation that similar optimal flux distributions were attained and strategies were clustered around a certain set of genes or pathways. The clustering of knockout candidates indicated the importance of fructose metabolism on levan biosynthesis, since all six genes associated with this pathway in the metabolic model were observed in the knockout strategies with an average observation frequency of 27 strategies per gene. The knockout genes were mainly associated with central carbon metabolism, purine metabolism, nitrogen metabolism, and metabolism of several amino acids (Table 3).
PAG, pathway-associated gene; GK/OS, gene knockout strategies; TCA, tricarboxylic acid.
Among the enzyme-coding genes, which have the repetition frequency higher than 3% in gene knockout strategies (Table 4), the knockout of the gene associated with the PTS system of fructose uptake seemed to be the most promising strategy for levan overproduction with 30% repetition in the optimal strategies. Therefore, we further analyzed the molecular components of the fructose uptake system to develop an optimal metabolic engineering strategy to enhance levan biosynthesis through studying nucleotide and amino acid sequences of fructose uptake system components.
PTS, phosphotransferase system; EC, Enzyme Commission.
PTSfru catalyzes the phosphorylation of fructose substrate during its translocation (Fig. 1). It is composed of FruA and FruB subunits. FruB is a fusion of fructose-specific enzyme II-A (EIIAFru), HPr, and enzyme I (EI) modules into a single polypeptide, and is responsible for conveying high-energy phosphates from phosphoenolpyruvate (PEP) into the system (Chavarría et al., 2012). The EIIAfru is a cytoplasmic enzyme and essential for the uptake of carbohydrate molecules through the cytoplasmic membrane and their phosphorylation. HPr is a central component of the bacterial phosphoenolpyruvate sugar PTS. The EI transfers the phosphoryl group from PEP to HPr across the cell membrane. HPr then transfers the phosphoryl group to one of several sugar-specific phosphoprotein intermediates. The conserved histidine in the N-terminus of HPr serves as an acceptor for the FruA and transports extracellular fructose through a phosphorylation-dependent process to yield fructose 1-P, which is then channeled to the central metabolism to yield fructose 1,6-bisphosphate.
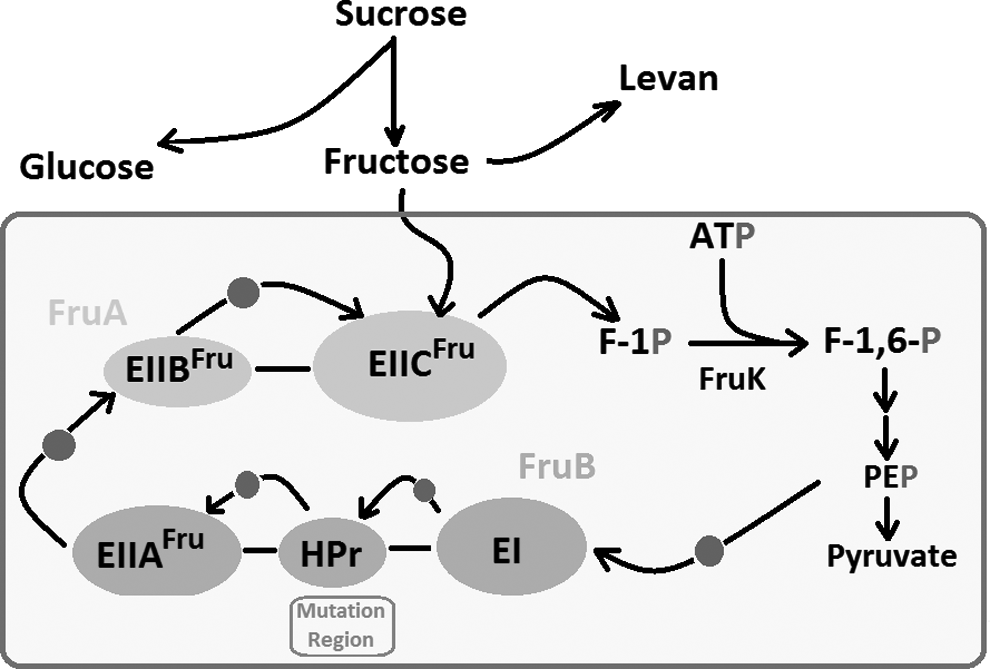
Proposed schematic representation of PTSfru system, levan production, and mutation region in Halomonas smyrnensis AAD6T (adapted from Chavarría et al., 2012). The red circle represents the transferred phosphate molecule. PTSfru, fructose-specific phosphotransferase system.
Knockout of HPr gene region in H. smyrnensis AAD6T
Considering its central role in the uptake system, HPr was selected as target gene region and insertion area for genetic manipulations. The knockout strategy was constituted by three subsequent steps: construction of pJet/HPr4 vector that carries the HPr region, which was then used for construction of pΩHOM2 by insertion of Ω cassette. Third construction was carried out by transferring HPr::Ω cassette into a suicide vector. The final product was used as donor strain for conjugal mating with target organism AAD6T by a helper plasmid (Fig. 2).
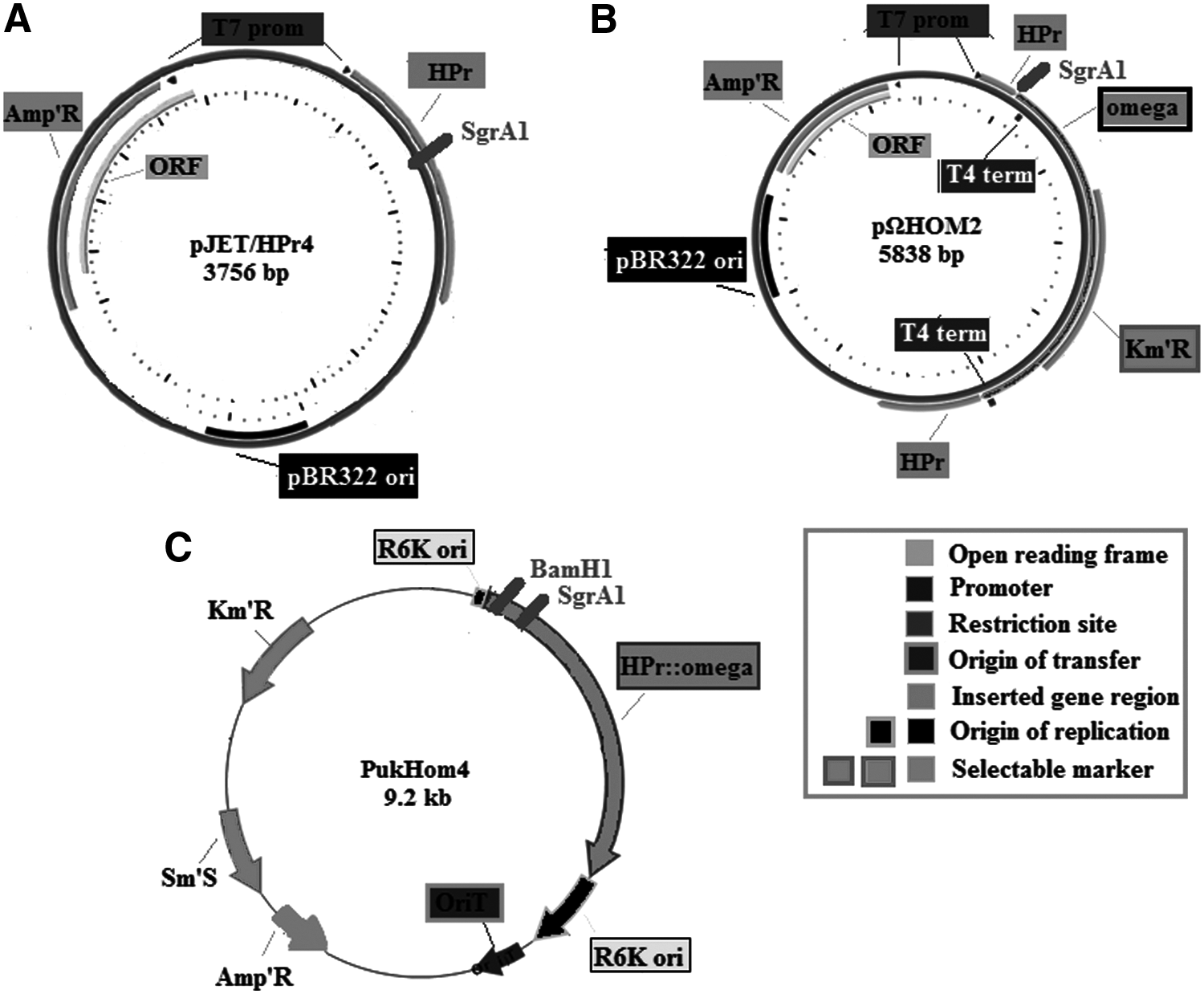
Plasmid vector maps of
After completion of desired construction that had HPr::Ω cassette on a suicide plasmid, the next step was the transformation of this region into recipient AAD6T. For transmission of PukHOM4 plasmid DNA into AAD6T genome, bacterial conjugation by triparental mating method (Vargas et al., 1997) was employed. It was composed of donor strain, PukHOM4, containing mobilizable plasmid of interest, and helper strain pRK2013 that has conjugative plasmid, which can mobilize the desired plasmid into recipient strain, AAD6T. The insertion of Ω cassette into the target HPr region was the crucial part of this study, since the whole work design was based on this insertional mutagenesis. Expected HPr::omega band was observed in mutant AAD6T (BMA14) by PCR amplification. HPr region in first construct (pJET/HPr4), and HPr::omega cassette in second construct (pΩHOM2) and transconjugants that carry HPr::omega cassette were confirmed by gel electrophoresis (Fig. 3) and sequencing.

Results of PCR amplifications:
Antibiotic susceptibility tests were performed to determine the antibiotic resistance of strains. Wild-type strain, AAD6T, has resistance for Km till 50 μg/mL, but showed no resistance to Amp and Str. Eventually, selective media were designed individually for each strain, and the minimal inhibitory concentration of the antibiotics in the media was determined (Table 5) to design media to confirm success of conjugation process.
Amp, ampicillin; Km, kanamycin; Str streptomycin.
Colony formation experiments indicated that conjugations were performed successfully, since growth was observed in SW-2 media supplemented with 100 μg/mL Amp, 100 μg/mL Km, and 50 μg/mL Str for BMA14 strain, whereas no growth was observed for AAD6T strain in optimum media supplemented with the antibiotics. In addition, production of levan in mucoid and slimy colonies was observed on plates with conjugated cells, as expected (data not shown).
Comparison of fermentation profiles AAD6T and BMA14 strains
When growth profiles of AAD6T and BMA14 strains were compared, significant differences were observed in growth rates and final biomass concentrations. BMA14 and AAD6T have growth rate values as 0.0043 h−1 and 0.0129 h−1, respectively (with R2 values 0.9708 and 0.9843 for BMA14 and AAD6T, respectively). Biomass concentrations at the end of 154 h were 2.38 and 4.54 g/L for BMA14 and AAD6T. On the other hand, no delay was observed in adaptation period of the mutant strain (Fig. 4A).
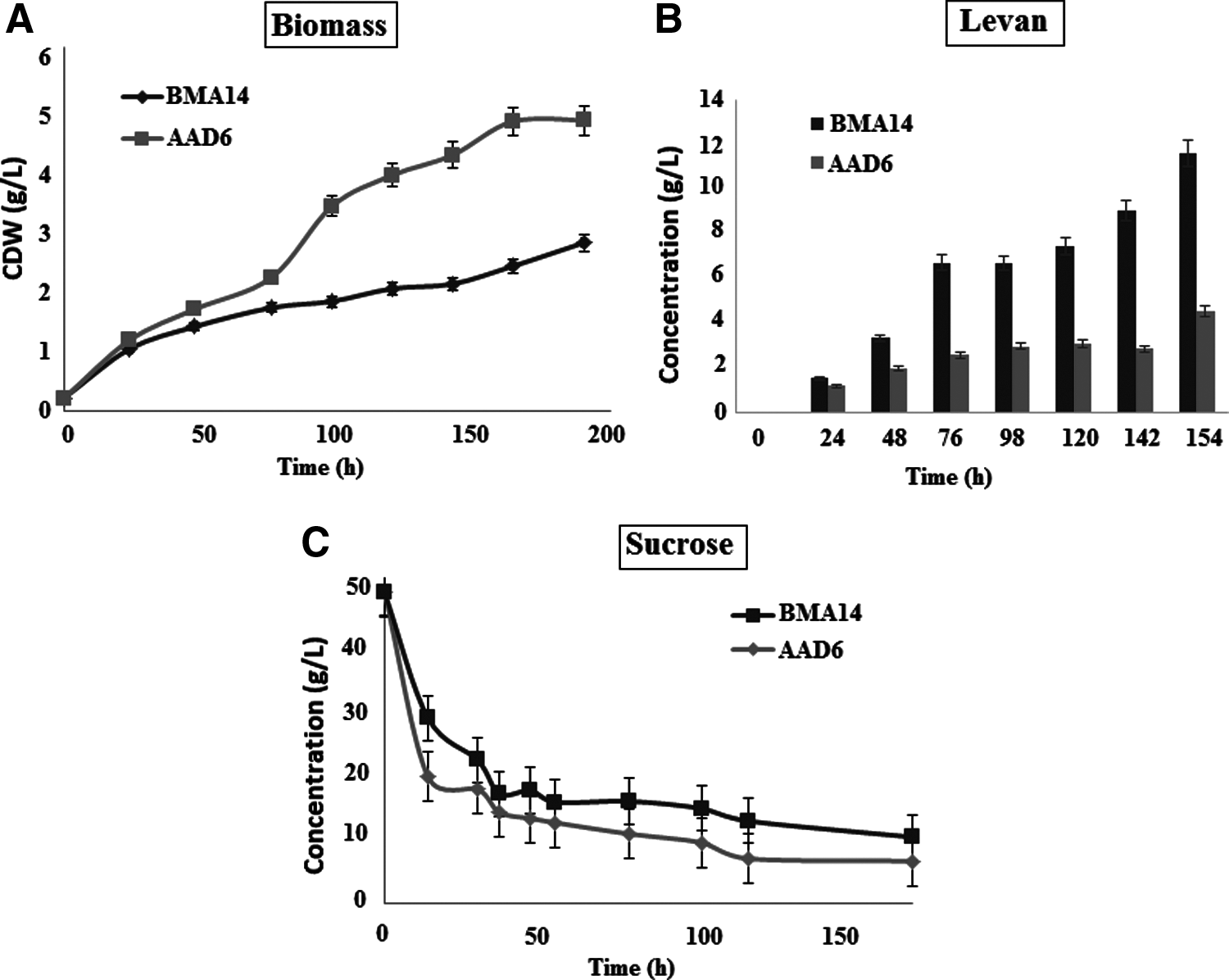
Cell growth
BMA14 strain exhibited enhanced levan production rates when compared to the wild-type strain. The final levan concentrations were 11.57 and 4.54 g/L in BMA14 and AAD6T, respectively. In addition, the product yield coefficient (YP/X) for levan (mass of levan per unit mass of biomass formed) was 4.86, whereas it was determined as 1.00 for the wild-type strain (Fig. 4B)
Differences between sucrose consumption profiles in AAD6T and BMA14 were analyzed through measuring extracellular sucrose in the growth media by HPLC (Fig. 4C). At the end of 154 h, sucrose consumption was 37.47 g/L for BMA14 and 41.14 g/L for AAD6T. Yield coefficient for substrate consumption per unit mass of biomass formation (YS/X) was determined as 15.75 and 9.06 in BMA14 and AAD6T, respectively.
As a result of low sucrose consumption rate and high yield coefficient for substrate consumption in BMA14, the conversion efficiency of sucrose into levan was significantly higher in BMA14 when compared to AAD6T (Table 6). In addition, theoretical carbon recovery calculation indicated that each mole of substrate (C12H22O11) was converted into 1.073 moles of biomass (CH0.66N0.2O0.3), 0.651 moles of levan monomer (C6H10O5), and 7.018 moles of CO2. Glucose assays were performed to measure extracellular glucose concentrations in both media; however, there was no significant difference between glucose consumption profiles of BMA14 and AAD6T (results were not shown).
All data were presented as mean values ± standard errors of the means.
Sucrose conversion efficiency = levan concentration (g/L)/consumed sucrose (g/L).
Discussion
Considering the widespread biotechnological production of EPSs, levan production capacities of various species have been investigated (Oner et al., 2016). In addition, several reports have been published concerning genetic and metabolic engineering of these species with the aim of levan overproduction (Feng et al., 2015; Fu et al., 2014; Moussa et al., 2017). This study aimed to show that levan producing H. smyrnensis AAD6T is an ideal platform for the development of new mutant strains with enhanced levan production capacity. For this purpose, using genetic and metabolic engineering techniques, a novel strain, BMA14, carrying the Ω cassette insertion in HPr region of the PTSfru, was developed and it showed enhanced capability in sucrose conversion efficiency and levan overproduction when compared to the wild-type strain, AAD6T.
To determine possible strain improvement strategies, we performed in silico knockout simulations for levan overproduction using the genome-scale metabolic model of H. smyrnensis AAD6T and a multiobjective optimization framework. In literature, several objective functions were employed to simulate bacterial growth. Since levan biosynthesis by AAD6T is growth associated, objective functions such as “maximizing biomass” or “maximizing levan production rate” would not be appropriate alone. Therefore, four different objective functions (BPCY, BPCY-BPCY, YIELD, and YIELD-YIELD), which seem to be appropriate for growth-associated levan biosynthesis, were employed in simulations with the ultimate purpose of identifying genetic modifications for levan overproduction.
Among those objective functions, the utilization of BPCY-BPCY, which represents the simultaneous maximization of growth rate and levan production rate, outperformed the others by presenting 100% of the gene knockout strategies identified by BPCY, 91% of the strategies identified through YIELD, and 84% of the strategies obtained through YIELD-YIELD. On the other hand, when the simulation results were comparatively analyzed, no significant differences were obtained among the optimization algorithms (SA, EA, and SPEA2), since 95% of all gene knockout strategies were identified by any of the algorithms.
The gene knockout strategies obtained through simulations with the utilization of different instruments (i.e., optimization algorithms and objective functions) were merged together and duplicates were removed. The comparative analysis of these knockout strategies indicated the importance of fructose metabolism on levan biosynthesis (Table 3) and presented that the knockout of the gene mtlA (EC 2.7.1.191) associated with the PTS system of fructose uptake seemed to be the most promising strategy for levan overproduction (Table 4).
The association of fructose uptake mechanism with levan biosynthesis is not surprising, since levan biosynthesis depends on utilization of the fructose moiety of sucrose at the extracellular environment, where the primary sucrose acts as a fructose acceptor to start chain lengthening, leaving a glucose moiety remaining at one end of the polymer. Therefore, we further analyzed the molecular components of the fructose uptake system and developed a metabolic engineering strategy on the insertion in HPr region of the PTSfru.
In the development of efficient biotechnological production schemes for levan producing microbial strains, the major bottleneck was the low rates of substrate consumption and product formation. In many bacteria, as Kotrba et al. (2001) suggest, a wide range of carbohydrates may be transported into bacterial intracellular environment by the PEP-dependent PTS, which assures optimum utilization of carbohydrates in complex environments. In H. smyrnensis AAD6T, PEP-dependent PTSfru is the essential fructose transfer pathway, which has pivotal roles in both substrate assimilation and metabolism. If phosphorylation of fructose was restrained by the downregulation of HPr, which is central component of the FruB, the utilization of the fructose moiety of sucrose might be depressed.
There are many studies showing the effect of PTS system and carbohydrate uptake mechanism on bioproduct formation, including biopolymers. It was reported that liberated fructose was metabolized by PTSfru, independent of the fructokinases in gram-negative bacteria such as Mannheimia succiniciproducens (Lee et al., 2010). Similarly, inactivation of HPr-ptsH gene resulted in remarkably high succinic acid yield in E. coli due to the significant alterations in carbohydrate uptake and metabolism (Liang et al., 2015).
In this study, genetic manipulation on HPr component of PTSfru was carried out by knockout target gene by inserting an antibiotic-resistant cassette (the Ω-Km fragment), which was successfully employed in genetic manipulations in halophilic bacteria such as H. elongata and H. eurihalina in previous studies (Cánovas et al., 1998; Göller et al., 1998; Llamas et al., 2000). Achievement of insertion into the host strain, AAD6T, in correct orientation was confirmed through colony formation on antibiotic-aided plates, PCR amplifications of insertion, and gel electrophoresis and sequencing. At the end of validations, the resultant mutant strain was named as BMA14.
Fermentation experiments indicated the high efficiency of the mutant strain BMA14 in terms of all production parameters analyzed (i.e., final levan concentration, sucrose consumption, sucrose conversion efficiency, and yield coefficients), when compared to the wild-type strain, AAD6T (Fig. 5). In previous studies, several stimulators such as mannitol (Ates et al., 2013) and boric acid (Sarilmiser et al., 2015) were reported for enhanced levan production in AAD6T with final levan concentrations of 6.60 and 8.84 g/L, respectively. The levan production was further improved with the development of BMA14, where the final levan concentration is elevated up to 11.57 g/L under the same fermentation conditions and growth medium, without any additional stimulators.

Biomass, substrate consumption, levan production, and yield comparison of mutant and wild-type strains (*represents values <0.05, ***represents values <0.001).
One of the major disadvantages of bioprocesses with mutant strains is the elongated adaptation periods in parallel with the decrease in productivity. On the other hand, the adaptation period of the mutant strain, BMA14, was not different from that of AAD6T; as a consequence, the fermentation period was not affected. Even though the growth rate of HPrfru mutant BMA14 was less than AAD6T, levan production by BMA14 was more effective than AAD6T with respect to higher final levan concentration and product yield coefficient (YP/X). In terms of sucrose conversion efficiency (i.e., g of levan produced per g of sucrose consumed), BMA14 displayed an efficient profile, almost threefold (Table 6).
The idea that relies behind the elevation of levan biosynthesis is based on increment in the assimilation of sucrose and utilization of the fructose moiety of substrate. Levan is synthesized by levansucrase enzyme (EC 2.4.1.10), which is a fructosyltransferase and typically able to catalyze two main reactions: (1) transglycosylation, employing the growing fructan chain (presence of polymerization), sucrose, or glucosaccharides and fructosaccharides (in oligosaccharide synthesis) as the acceptor substrate; (2) and sucrose hydrolysis, using water as the acceptor.
In our case, fructosyltransferase acts as a vehicle for transferring fructosyl residues of sucrose to a variety of acceptors. The disruption of the HPr component of PTSfru could lead to reduced metabolic overflow (i.e., for cell maintenance) and might boost the transfructosylation process, which culminates in high level of sucrose assimilation and substrate conversion efficiency. An accretion in the conversion of sucrose might end up with the increased activity of levansucrase, and sucrose is catabolized and polymerized into fructan chain more effectively. As a consequence, levan biosynthesis is improved. Restructuring of PTS system HPrfru component was accomplished with the most effective way of levan production from a halophile, reported till now.
Conclusions
Metabolic systems engineering is being used to redirect microbial metabolism for the overproduction of chemicals of interest with the aim of transforming microbial hosts into cellular factories. In this study, H. smyrnensis AAD6T is considered a candidate microbial cell factory for systems metabolic engineering studies toward the development of novel strains with improved levan biosynthesis. Through OptFlux simulation platform and utilization of different optimization algorithms and objective functions, simulations were performed in silico to determine optimal gene knockout strategies to develop new strains with enhanced levan production capability. The coupling of BPCY-BPCY objective function with SPEA2 algorithm seemed to be the most efficient instrument combination to identify gene knockout strategies in our case study.
The gene knockout strategies emphasized the vital role of the fructose uptake mechanism in levan biosynthesis, and the PTSfru was proposed as the most promising target for further metabolic engineering studies. Then, a new metabolically engineered H. smyrnensis strain, BMA14, with enhanced levan production capability was developed through carrying out insertional mutagenesis and triparental mating techniques in HPr region of the PTSfru system. The high efficiency of the mutant strain BMA14 in terms of final levan concentration, sucrose consumption rate, and sucrose conversion efficiency was demonstrated through fermentation experiments.
As far as we know, this work is the first report of a systematical metabolic engineering approach with elevated levan production in a Halomonas strain. The genome-based metabolic systems engineering approach presented in this study might be considered an efficient framework to redirect microbial metabolism for the overproduction of chemicals of interest, and the novel strain BMA14 might be considered a potential microbial cell factory for further studies aimed to design levan production processes with lower production costs.
Footnotes
Acknowledgments
This work was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) through 110M613 and Marmara University Research Fund (BAPKO) through FEN-C-YLP-060911-0280, FEN-C-YLP-101013-0404, and FEN-A-130511-0163 projects.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.