
Abstract
Abstract
Neurology research and clinical practice are transforming toward postgenomics integrative biology. One such example is the study of human brain metabolism that is highly sophisticated due to reactions occurring in and between the astrocytes and neurons. Because of the inherent difficulty of performing experimental studies in human brain, metabolic network modeling has grown in importance to decipher the contribution of brain metabolite kinetics to human health and disease. Multiomics system science-driven metabolic models, using genome-scale and transcriptomics Big Data, offer the promise of new insights on metabolic networks in human brain. Added to this, the availability of omics technologies in both developed and developing world, neurology research, and clinical practice ought to be repositioned with a view to systems medicine. In this expert analysis, we present a critical and in-depth overview of the basic tenets of human brain metabolism, together with the most recent metabolic modeling strategies and computational studies of brain in health and neurological diseases. Human genome-scale metabolic models developed in a both global and brain-specific manner and multiomics synthesis of knowledge are highlighted in particular. We conclude by underscoring the value of multiomics modeling for metabolic diseases and computational investigations of the brain networks, with a view to unlocking the pathophysiology of Alzheimer's disease, Parkinson's disease, migraine, stroke, epilepsy, and multiple sclerosis, among other neurological disorders of importance for global health.
Introduction
N
ALS, amyotrophic lateral sclerosis; HD, Huntington's disease; MS, multiple sclerosis.
Nervous system is divided into two main parts as the central nervous system (CNS) and the peripheral nervous system (PNS) (Mai and Paxinos, 2011). The CNS is made up of the brain and spinal cord. Signal transmissions and metabolic reactions in the nervous system are considerably complicated due to high level of interactions (Myers and Olson, 2012). Because of these complexities of the brain network, any perturbation or abnormal development in the nervous system may result in different metabolic outcomes and neurological disorders whose molecular mechanisms have not been fully understood.
Human metabolism comprises thousands of biochemical reactions taking place in the human cells (Brunk et al., 2018). This set of reactions gives rise to substantially sophisticated metabolic networks due to the presence of a single metabolite in various metabolic processes. Among all, the human brain is the most complex organ due to its reactions occurring in and between the astrocytes and neurons (Allaman et al., 2011; Belanger et al., 2011).
Because of the inherent difficulty of performing experimental studies on metabolic changes in the human brain, metabolic network modeling plays an indispensable role to decipher the contribution of its metabolite kinetics at health and disease (Lewis et al., 2010; Martin-Jimenez et al., 2017; Sertbas et al., 2014). In accordance with this, not only generic human genome-scale metabolic models but brain-specific networks have also been reconstructed for elaborated investigations via systems biology approach.
Recent cutting edge developments in multiomics data integration and technologies (Horgan and Kenny, 2011), encapsulating genomics (Macaulay and Voet, 2014), transcriptomics (Lowe et al., 2017), proteomics (Aslam et al., 2017), and metabolomics (Liu et al., 2014) have lead to new insights in decoding of metabolic networks in human tissues as well as in various organisms (Heavner and Price, 2015; McCloskey et al., 2013; Yilmaz and Walhout, 2017). Added to this, the availability of omics technologies in both developed and developing world (Mitropoulos et al., 2017) and neurology research ought to be repositioned with a view to systems medicine (Gollapalli et al., 2017; Oliveira et al., 2017).
Omics technologies provide researchers with an opportunity of simultaneous analysis of thousands of gene expression, protein, and metabolite levels in a single microarray experiment. Integration of data and research by multiomics studies reveals molecular interactions among the genome, metabolome, and proteome (Chubukov et al., 2014; Piazza et al., 2018). Recently, a taxonomy for multiomics studies was also proposed, underscoring the growing importance of such postgenomics integrative approaches to understand the systems biology of human health and disease (Pirih and Kunej, 2017). Consequently, the quality of metabolic networks, including the number of reactions, metabolites, and genes, covered in genome-scale metabolic models, drastically increased with integrative multiomics analyses (Geng and Nielsen, 2017).
The present expert review underscores that the field of neurology is increasingly moving toward postgenomics integrative approaches. The review first presents a clinical contextual presentation of various neurological diseases, followed by multiomics and metabolic modeling approaches that can inform neurology research, and in due course, clinical practice in the future.
Clinical Context for Neurological Disease and Phenotypes
Dementia states a general term used for decreased mental ability, which is caused by the damage of nerve cells in the brain and affects daily life severely. Its symptoms include memory loss, anxiety, depression, and difficulties in communication, reasoning, and complex tasks. The most frequent type of dementia is Alzheimer's disease (AD) accounting for about 60% to 80% of dementia cases (Alzheimer's Association, 2017; Nussbaum and Ellis, 2003). It is pathologically characterized by accumulation of amyloid β-peptide outside neurons and tau tangles inside neurons (Alzheimer's Association, 2017; Mawuenyega et al., 2010).
The most important risk factors for AD are age, family history, and genetic factors. Nevertheless, sporadic cases cover more than 90% of AD cases (Bekris et al., 2010). APOE gene is considered to be linked with the sporadic late onset of AD and the mutations in three candidate genes in particular (βAPP, PSEN1, PSEN2); the Down syndrome plays role in early onset of this disease as well (Table 2). In the treatment of the AD, acetylcholinesterase inhibitors and N-methyl D-aspartate (NMDA) receptor antagonists are recommended to improve cognition, behavior, and functional state, but robust therapeutics innovation is still very much needed (Mangialasche et al., 2010).
The most widely encountered.
NMDA, N-methyl D-aspartate.
In a context of other common neurological disorders without an established pathophysiology, migraine and epilepsy are common, paroxysmal, chronic, and strongly associated neurological diseases stemming from electrical disturbances in the brain (Nye and Thadani, 2015). Migraine is characterized by attacks, including severe throbbing pain and light and sound sensitivity, due to neuronal overactivity. Depending on the frequency of headache per month, migraine is classified as chronic (less than 15 days/month) and episodic (15 days or more/month) (Katsarava et al., 2012). Age, low socioeconomic status, head injury, attack frequency, medication overuse, obesity, stress, and caffeine overuse are some of the notable risk factors for migraine progression (Bigal and Lipton, 2006).
In epilepsy, brain is disrupted by excessive electrical activity leading to an epileptic seizure, which may be stimulated by migraine. It affects about 70 million people from all age groups around the world (Ngugi et al., 2010). According to twin studies (Kjeldsen et al., 2003), hereditary and individual-specific factors constitute about 80% and 20% of epilepsy, respectively.
Another notable neurological disease commonly observed among the world population is Parkinson's disease (PD), which occurs as a result of dysfunction of dopaminergic neurons in the substantia nigra pars compacta with accumulation of the misfolded proteins (Dauer and Przedborski, 2003; Nussbaum and Ellis, 2003). Loss of these dopamine-producing neurons results in the decline in the level of dopamine neurotransmitter, which takes part in the regulation of movement and cognition (Sasidharakurup et al., 2017).
Sporadic cases constitute nearly 95% of the PD cases, the rest are inherited. Mutations in genes, including α-synuclein, parkin, UCH-L1, and DJ-1 (Table 2) can cause emergence of parkinsonism (Ross and Pickart, 2004). In the treatment of a patient with PD, oral administration of levodopa, which is the precursor of dopamine, is a widely used strategy (Connolly and Lang, 2014; Cools, 2006).
For the biologically proper functioning of a protein, posttranslational modifications, including protein folding, are a requisite. Huntington's disease (HD) is an inherited neurological disease resulting from increased number of cytosine, adenine and guanine (CAG) trinucleotide repeats in the huntingtin gene (Ross and Tabrizi, 2011). The repeated glutamine amino acids lead to misfolding and aggregation of huntingtin protein and cause neuronal metabolic interruptions as well as cell death (Pringsheim et al., 2012). The diagnosis of HD is determined by family history, genetic testing, and the progress in symptoms, including chorea, behavioral features, and dementia. Due to the unavailability of robust treatments to change the course of HD, symptom-oriented treatments are customized to enhance patient's life quality (Adam and Jankovic, 2008).
Myelin sheath is a protective layer coating nerve fibers for proper functioning of the nervous system. The damage of this insulating layer by immune system cells gives rise to impairment of nerve impulse transmission and the neurological disorder multiple sclerosis (MS), affecting more than 2 million people worldwide (Browne et al., 2014; Frohman et al., 2006). MS is diagnosed by McDonald Criteria based on clinical attack and magnetic resonance imaging for dissemination of lesions in space and time (Polman et al., 2011). The most common symptoms of MS are fatigue, numbness, spasticity, and problems in vision, bowel, and bladder. Genetic and environmental factors are the causes of MS (Compston and Coles, 2008). Familial cases constitute about 20% of MS cases.
Progressive degeneration of the motor neurons brings about amyotrophic lateral sclerosis (ALS) that has higher incidence among men compared with women population (Kiernan et al., 2011; Logroscino et al., 2010). Motor neurons are responsible for important muscle activities; therefore, this neurodegenerative disease causes muscle-related problems covering walking, speaking, and swallowing. The underlying causes of ALS are genetics, epidemiologic features, exposure to heavy metals, viral infection, and prion disease (Rowland and Shneider, 2001). Approximately 10% of ALS cases are familial, and the rest is sporadic. The diagnosis is performed by electrodiagnostic, neurophysiological, neuroimaging, and clinical laboratory studies (Brooks et al., 2009).
Insufficient supply of blood to the brain results in impairment of brain metabolism. This neurodegenerative state is called stroke, where nutrients and oxygen are depleted, eventually leading brain cells to die. Diabetes, high blood pressure, smoking, high cholesterol, obesity, stress, and lack of physical activity are the common risk factors for stroke (Hickey et al., 2009). The warning signs of this fatal disease include sudden numbness or weakness especially on one side of the body, sudden confusion or speaking difficulty, sudden vision problem, dizziness or loss of balance, or sudden and severe headache of no known cause. Stroke has globally resulted in about 6.5 million deaths in 2013; it was the second leading cause of death after ischemic heart diseases at that time (Benjamin et al., 2017).
In addition to aforementioned neurological diseases, other common neurological disorders include autism spectrum disorder (ASD), cerebral palsy (CP), Tourette syndrome (TS), traumatic brain injury (TBI), and spinal cord injury (SCI) (Hirtz et al., 2007). ASD, CP, and TS are the neurological disorders with an onset early in life. ASD is the neurological condition which brings about problems in communication skills (Elsabbagh et al., 2012).
CP is resulted from the nonprogressive damage in the brain under development and leads to physical impairments, including body movement and muscle coordination (Oskoui et al., 2013). TS is neuropsychiatric disorder diagnosed by existence of motor and vocal tics (Scharf et al., 2015). TBI and SCI are the neurologic disorders with onset at any age, and they are mainly caused by blow or jolt to the head or spinal cord (Langlois et al., 2006; Roozenbeek et al., 2013; Singh et al., 2014).
The age of onset of different neurological diseases demonstrate the variability throughout the life span (Hirtz et al., 2007). Despite the rare cases before the age of 65, AD and PD are the late-onset diseases mostly seen in the population aged 65 years and older, and their prevalence increases with age (de Lau and Breteler, 2006; Hebert et al., 2013; Nussbaum and Ellis, 2003). Onset of ALS goes up after the age of 40 years and is highest between 70 and 74 years (Logroscino et al., 2010). The average age of onset for HD is also 40 with a very broad range of age between 3 and 85 years (Myers, 2004; Ross and Tabrizi, 2011).
The mean age of stroke onset is about 70 years; however, it affects all age groups with the rare cases in children (Rowe et al., 2013; Traylor et al., 2015). The incidence of epilepsy is higher in children and elderly people than those in adults (Bergey, 2004). Migraine onset can be seen at all ages, the most reported cases being among adults (Jensen and Stovner, 2008). For MS, the mean age of the onset is about 32 (George et al., 2016).
Nervous system is a complex network, which has potential for computational modeling studies (Gruetter et al., 2001; Sterratt et al., 2011). The content and accuracy of these models increased in line with the developments in neuroscience and computer science. In the last two decades, systems biology has emerged as the field of study, focusing on system-level understanding of biological systems with the help of computational approaches (Chuang et al., 2010; Kitano, 2002).
Applications of this field of science to nervous system allow us to decipher the molecular mechanisms behind the various neurological states, including aforementioned diseases. An important part of the systems biology research in human metabolism is the genome-scale metabolic models, which include metabolite, reaction, and associated gene information.
Human Genome-Scale Metabolic Models
Since the past decade, extensive biological evidence was utilized in reconstruction of the first genome-scale human metabolic network (Recon1) modeled at high level of detail and accuracy (Duarte et al., 2007). The latter comprehensive manual literature survey provided classification of the metabolic pathways into three categories:
1. Pathways in which almost all enzymes and their related genes are known, 2. Pathways having moderate literature support, and 3. Pathways incompletely elucidated and required to be investigated further.
By the approach of human genome-scale metabolic modeling in the study by Duarte et al. in 2007, the gaps in the network was identified and targeted studies were carried out for the missing metabolic reactions. A bottom-up approach is applied in the manual reconstruction of high-quality human metabolic networks, which represent extracellular medium and seven compartments, including cytoplasm, mitochondria, nucleus, endoplasmic reticulum, Golgi apparatus, lysosome, and peroxisome.
The validation of the basic functionality of Recon1 was performed by simulating 288 known metabolic functions in silico (Duarte et al., 2007). The primary advantage of Recon1 is a careful formulation of literature based human-specific data, including metabolites, reactions, directionality, gene, and protein information. This enables Recon1 to integrate with omics technology to decipher changes in the metabolic network due to any kind of perturbations, including diseases or drug treatment.
In the same year (in 2007), the second human-specific genome-based metabolic network (Ma et al., 2007), starting from human gene and enzyme annotation information from different databases, including KEGG (Kanehisa et al., 2004), Universal Protein Resource (UniProt) (Wu et al., 2006), HCNC (Eyre et al., 2006), NCBI Entrez Gene (Maglott et al., 2005), Ensembl (Hubbard et al., 2007), and GeneCards (Safran et al., 2002), was developed. An initial list of reactions, corresponding to related genes and enzymes, were generated from Uniprot and KEGG ligand database (Goto et al., 2002) and the reactions in the list were manually checked from the literature whether they exist in the human metabolism.
To come up with a more comprehensive human metabolic network, this initial reaction list was combined with EMP database (Enzymes and Metabolic Pathways), which contains a compound centric metabolic network specific to human based on literature. The final high-quality network, referred to as Edinburgh Human Metabolic Network (EHMN), includes 2823 reactions and 2322 genes (Fig. 1). Different from Recon1, EHMN does not cover subcellular location information, which considerably contributes to increase in the total number of the reactions in the developed metabolic network due to the repetition of the same reactions in different compartments.
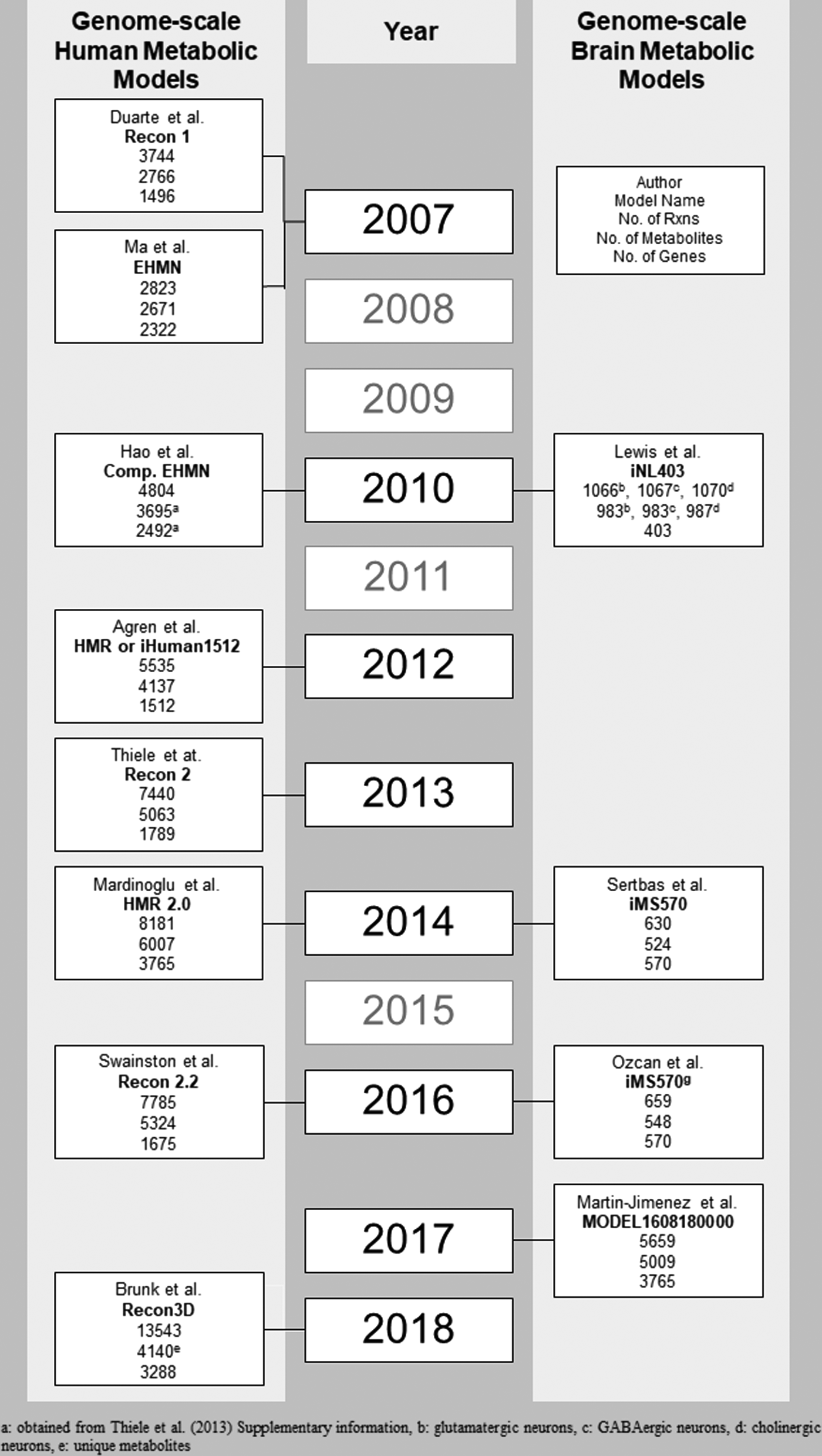
Chronological developments in genome-scale human metabolic networks and brain-specific metabolic networks.
The application of functional analysis to EHMN classifies the noncentral metabolites, which are not in glycolysis, pentose phosphate pathway (PPP), and tricarboxylic acid (TCA) cycle pathway, as exchangeable, degradable, synthesizable, and isolated similar to bow-tie structure (Ma and Zeng, 2003). In 2010, EHMN was extended with subcellular compartment information covering extracellular, nucleus, cytosol, endoplasmic reticulum, Golgi apparatus, peroxisome, lysosome, and mitochondria (Hao et al., 2010). Subcellular location information was extracted from Gene Ontology (Ashburner et al., 2000) and SWISS-PROT (Boeckmann et al., 2003) in the process of compartmentalization, which enhances the number of reaction to 4804 in the human metabolic network.
Agren et al. (2012) reconstructed Human Metabolic Reaction (HMR) database by integrating the information from previously developed human genome-scale metabolic networks and databases [Recon1, EHMN, HumanCyc (Romero et al., 2005), and KEGG]. HMR database, referred to as iHuman1512, includes 5535 metabolic reactions and 1512 genes (Fig. 1). Subsequent to the reconstruction of comprehensive HMR database, they proposed Integrative Network Inference for Tissues (INIT) algorithm, which describes integration of this generic database with Human Protein Atlas and gene expression data with the ultimate purpose of automatically generated tissue-specific network.
To evaluate the ability of their database and algorithm in tissue-specific metabolic network reconstruction, they generated hepatocyte-specific metabolic network (iHepatocyte1154), and it was compared with HepatoNet1 (Gille et al., 2010), which is a manually curated liver-specific genome-scale metabolic network available in the literature. They found 452 genes in common among 1154 and 713 genes in the iHepatocyte1154 and HepatoNet1 models, respectively. Then, to identify metabolic features of cancer, the reporter metabolite analysis (Patil and Nielsen, 2005) was performed, which revealed significant changes in polyamines metabolism, isoprenoid biosynthesis pathway, prostaglandins, and leukotrienes.
The key aim of the human genome-scale metabolic network studies is to reconstruct the most extensive model representing metabolic interactions occurring in human metabolism. In accordance with this purpose, substantial part of community, experienced in genome-scale models around the world, made a consensus to come up with the up-to-date global human metabolic model (Recon2) by using available human metabolic models at that time (Thiele et al., 2013). In addition to Recon1 (Duarte et al., 2007), the other models integrated to Recon2 are the compartmentalized EHMN (Hao et al., 2010), HepatoNet1 (Gille et al., 2010), Ac-FAO module (Sahoo et al., 2012), and hs_sIEC611 (Sahoo and Thiele, 2013).
The total number of the reactions (7440) and metabolites (5063) covered by Recon2 (Fig. 1) increased by approximately twofold in comparison to its predecessor Recon1. The biomarker predictive capabilities of Recon1 and Recon2 were compared for inborn errors of metabolism and found with accuracy of 63% and 77%, respectively. Recon2 is a global human genome-scale metabolic network and can be regarded as a template in the reconstruction of tissue-specific metabolic models for different tissues.
Consequently, 65 cell type-specific metabolic models were generated by using expression data from Human Protein Atlas (Uhlen et al., 2010) and published algorithms (Schellenberger et al., 2011; Shlomi et al., 2008). The results showed that 8% of the Recon2 reactions and 26% of the Recon2 genes appeared in all cell type-specific models. Thirty-three percent of the reactions and 14% of the genes were found in none of the cell type-specific models.
A new hepatocyte model generated from Recon2 was compared with the two previously published hepatocyte-specific models (Gille et al., 2010; Jerby et al., 2010). Around 1100 reactions of generated hepatocyte model (including 3041 total number of reactions) were represented in both Jerby at al. (2010) and HepatoNet1 (Gille et al., 2010), which are covering 1823 and 2539 reactions in total, respectively. The differences between these previous hepatocyte models and automatically generated model indicate the importance of the manual curation and assessment. Due to the high coverage of metabolic enzymes and enzymatic complexes in Recon2, they also mapped enzymes in Recon2 with DrugBank (Wishart et al., 2008), which involves drug-enzyme mapping information, providing approaches for drug action simulations in modeling studies.
An extensive lipid metabolism is not included in the aforementioned generic human networks. To decipher the alterations in lipids during the development of different diseases and their effects on molecular interactions in the human tissues, Mardinoglu et al. (2014) integrated comprehensive lipid metabolism with HMR database to generate HMR 2.0 (Fig. 1). The addition of lipid metabolism into genome-scale human metabolic network enabled in-depth investigation of the link between lipid metabolism and other cellular metabolism.
They reconstructed hepatocytes-specific genome-scale metabolic model (iHepatocytes2322) based on HMR 2.0 and proteomics data. Reporter metabolite and subnetwork analyses showing metabolites and pathways, which had the most significant transcriptional changes due to perturbations, were performed by using the new hepatocytes-specific model and transcriptomics data from healthy subjects and patients with nonalcoholic steatohepatitis. The results revealed that the level of chondroitin and heparan sulfates in the blood samples can be used as the biomarkers for the diagnosis of this disease. Their model-based study also pointed out mechanistic explanation of serine shortage in nonalcoholic steatohepatitis patients and suggested that phosphoserine phosphatase, serine hydroxymethyltransferase 1, and branched-chain amino acid transaminase 1 were the potential therapeutics for the treatment of this liver disease.
Several updates are available for Recon2 to improve the modeling competence, including carbon balancing (Smallbone, 2013), drug metabolism (Sahoo et al., 2015), protein transport (Sahoo et al., 2014), and error corrections (Quek et al., 2014). These updates were compiled in Recon2.2 (Swainston et al., 2016) with the several developments, which cover comprehensive manual curation, mass and charge balance of reactions, and extended energy generation. The updated genome-scale human metabolic network (Recon2.2) has 7785 reactions, 5324 metabolites, and 1675 genes (Fig. 1).
To compare Recon2.2 with the previous versions, the maximum ATP yields per unit carbon source were computed by using the individual human metabolic models. Among them, Recon2.2 was able to estimate maximum ATP fluxes in accordance with the theoretical yields.
The most recent human genome-scale network is Recon3D (Brunk et al., 2018), which was reconstructed by extending Recon2 and integrating reactions from different sources, including HMR 2.0, drug module (Sahoo et al., 2015), transport module (Sahoo et al., 2014), host-microbe reactions (Heinken and Thiele, 2015), and dietary compounds metabolism. Different from the previous human genome-scale networks, Recon3D covers three-dimensional structure information for metabolites and proteins, in addition to high number of reaction, metabolite, and gene content (Fig. 1). All of its reactions are manually curated to delete unneeded reactions.
Recon3D was used in transcriptome analysis of drug data to elucidate perturbed metabolic pathways via machine-learning approach. The analysis of 47 drug indications demonstrated that indication-specific drugs cause similar gene expression pattern. For many drugs, the metabolic responses were greatly conserved and this conservation was especially high for the antipsychotic drugs. The computation of the drug response by using protein and metabolite structural data from Recon3D pointed out the structurally different molecules, which fulfil similar effects on metabolic pathways.
Tissue Specific Genome-Scale Networks
A “must” of multiomics modeling
In accordance with the perpetual developments in generic networks mentioned above, several context-specific genome-scale metabolic networks were reconstructed for different kinds of human tissues, including erythrocyte (Bordbar et al., 2011), liver (Gille et al., 2010; Jerby et al., 2010), kidney (Chang et al., 2010), myocytes (Varemo et al., 2015), and brain (Lewis et al., 2010; Martin-Jimenez et al., 2017; Sertbas et al., 2014). These continual enhancements provided an elaborated understanding of network-centric molecular mechanism via systems biology approach and multiomics technology. It is clear that the active pathways in various types of human tissues demonstrate differences due to the nature of the specific tissue.
Generic human networks are essential elements in the reconstruction of tissue-specific genome-scale human metabolic network. If a functioning pathway is unavailable in the generic network, which is selected as template in the tissue-specific reconstruction, it is not possible to capture this active pathway in the developed cell type-specific network without conducting time-consuming literature survey. Therefore, it is suggested that a well-established generic human network should cover all the metabolic reactions, pathways, and other related information (metabolites, genes) existing in all specific human tissue. Undoubtedly, this contributes to a better whole systems understanding of underlying mechanism due to perturbations (diseases, drug actions).
Genome-Scale Human Brain Modeling
Initial attempt in modeling of brain metabolism was performed by Chatziioannou et al. (2003) and they included 16 metabolic reactions depending on interactions in and between the neurons and astrocytes. In 2007, two different metabolic models were reconstructed by Cakir et al. (2007) and Occhipinti et al. (2007), including 217 and 108 reactions, respectively. After the reconstruction of generic human metabolic network (Recon1), Lewis et al. (2010) developed a genome-scale human brain-specific metabolic model originated from Recon1 by using brain proteome databases and literature survey.
They reconstructed separate brain metabolic networks for glutamatergic, GABAergic, and cholinergic neurons, which provide an advantage with a close understanding of cellular mechanism in different type of neurons. Their model, depending on neuron type, involves around a thousand of reactions as well as compartment-specific metabolites (Fig. 1). The total number of the genes associated with these three models is 403; therefore, the model was named as iNL403. The validation of the model was performed by comparing the flux distribution results with the experimentally measured values reported in the literature.
Subsequent to building of their brain-specific genome-scale model, they explored the AD metabolism computationally so as to get new insights on putative genes. Experimental studies in the literature suggest that glutamatergic and cholinergic neurons are more sensitive to AD, and GABAergic neurons are not affected until later stages of AD. The iNL403 model was used to computationally investigate the potential genes causing these neuron-specific effects. Glutamate decarboxylase (GAD) has neuroprotective capacity, and neurons with low GAD expression are affected earlier than those with high GAD expression (Lai et al., 2007; Lewis et al., 2010).
Sertbas et al. (2014) reconstructed a new genome-scale brain-specific metabolic model (iMS570) originated from Cakir et al. (2007) by expanding lumped reactions into elementary reactions and adding new pathways after an extensive literature survey with the corresponding gene information available for each reaction. The resulting model contains 630 reactions, 524 metabolites, and 570 genes (Fig. 1). The brain model was validated such that the flux distribution, performed by flux balance analysis (FBA) in healthy condition, gives consistent values with the experimental studies. Then, the model was used for further investigation of transcriptome data for six different neurological diseases including, AD, PD, ALS, MS, HD, and schizophrenia.
Reporter metabolite analysis (Patil and Nielsen, 2005) and reporter pathway analysis, showing metabolites and pathways that have the most significant transcriptional changes due to perturbations, were carried out for six diseases and disease-specific reporter metabolites and pathways were found, respectively. Their computational outcomes demonstrated significant change in TCA cycle, oxidative stress, lipid metabolism, and many amino acid associated pathways under the condition of the studied diseases. For each reporter metabolite, computational binding site analysis was performed to identify overrepresented transcription factors for the six neurological diseases.
The transcription factors, including KLF4, USF1, SP1, and others from PAX, FOX, and Sox protein families, were found to be overrepresented, and they are common in more than one studied neurological disorders.
We, in this study, simulated the stroke effect by decreasing the uptake levels of the metabolites into the brain, so as to see glutamate change in the case of stroke via iMS570. The methods such as FBA and minimization of metabolic adjustment used in computation are described in detail elsewhere (Cakir et al., 2007; Sertbas et al., 2014). The predicted result from this modeling study (Fig. 2) is in agreement with the experimental findings where the glutamate level was measured during the changing cerebral blood flow due to reversible middle cerebral artery occlusion (Feustel et al., 2004).
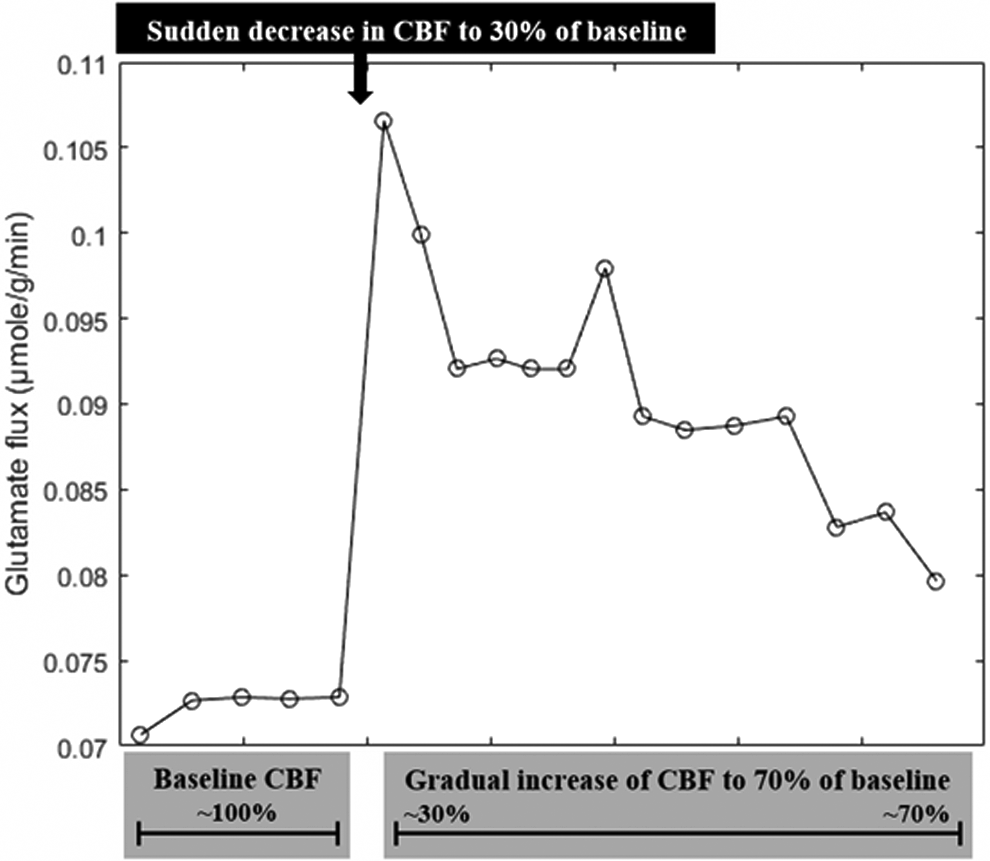
Computational prediction of glutamate level via iMS570 in case of stroke.
Not only human genome-scale metabolic networks unveil the variations in cells due to metabolic diseases but they also serve as a bridge between cancer progression and cellular mechanism in promising selective treatments as well (Folger et al., 2011). Glioblastoma is the most prevalent malignant brain tumor in the society. To compute the metabolic flux distributions in glioblastoma, Ozcan and Cakir (2016) extended iMS570 by inserting biomass growth reactions, representing tumor proliferation, to generate growth-implemented brain model, called iMS570g.
Two different computational methods, known as GIMME (Becker and Palsson, 2008) and MADE (Jensen and Papin, 2011) integrated iMS570g with glioblastoma transcriptome data to come up with glioblastoma-specific metabolic models as well as metabolic flux distributions. The results of this in silico glioblastoma metabolic model were in accordance with in vitro and in vivo studies in the literature. That glioblastoma-specific metabolic network offers potential implementations, including specific metabolic simulations and therapeutic applications. Under the condition of high rate of glucose consumption, all their computational results indicated high lactate production rate and relatively low TCA cycle activity, which are known as Warburg effect (Warburg, 1956).
Martin-Jimenez et al. (2017) reconstructed genome-scale astrocyte-specific metabolic network (Fig. 1) by using the comprehensive protocol (Thiele and Palsson, 2010). As an initial reconstruction, they used astrocyte omics data and Human Metabolic Atlas (Pornputtapong et al., 2015). Manual curation was applied to the initially generated network of the genes and their associated reactions to support their existence in the astrocytes by the literature and obtain computationally feasible astrocyte-specific metabolic model with no gaps and no dead-end metabolites.
The curated astrocyte model was transformed into a mathematical model, which can be solved by optimization techniques. FBA was performed to predict metabolic fluxes under normal (healthy) and ischemic conditions by gradually decreasing oxygen consumption and glucose intake. The comparison of flux distributions under ischemic condition with those under normal state demonstrated significant flux decline in TCA cycle, fatty acids oxidation, oxidative phosphorylation, and glutamate–glutamine cycle reactions and increase in PPPs as well as anaerobic glycolysis. Since the model is specific to astrocyte metabolism, it can be used in computational investigation of changes in astrocytes due to perturbations.
Conclusions
Postgenomic systems science innovations and multi-omics modeling to study human brain metabolite kinetics can usefully inform neurology, as an interdisciplinary field of inquiry. The proper functioning of the CNS and PNS necessitates a coordinated process of reactions occurring in and between nervous tissues. Any kind of perturbation, resulting in malfunction of these perfectly coordinated nervous cells system, gives rise to various neurological disorders, which is difficult to study experimentally; however, easy to simulate computationally.
Generic and brain-specific genome-scale human metabolic networks are notable tools that can inform neurology research and help interpret results obtained in experimental studies. The experimentally measured levels of certain metabolites are defined as the constraints to brain metabolic model to figure out the distribution of all metabolites in the metabolic network. The use of these computational approaches together with multiomics discloses how metabolic levels and pathways are affected exhaustively due to a specific perturbation or disease state.
Further developments of these networks should enable researchers to decipher the underlying molecular interactions in the nervous system due to the diseases and to come up with promising therapeutic suggestions. Their integrations with the multiomics technologies offer great potential in the future development of systems medicine and personalized medicine. Most of the current neurological treatments regard a given patient as an average patient. However, it is not possible to fit an average treatment, and this may result in an ineffective treatment.
Multiomics technologies reveal the detailed features of person-specific metabolism, and their integration with the genome-scale metabolic model can recommend personalized or precision medicines. Undoubtedly, simulating the interactions among multiple tissues in multiple organisms, genome-scale metabolic models can be successfully applied for developing individualized therapies and diagnostics in neurology in the future.
Footnotes
Acknowledgments
The authors gratefully acknowledge the intramural Bogazici University Research Fund (Project 111 M41).
Author Disclosure Statement
The authors declare that no competing financial interests exist.