
Abstract
Abstract
Spinal muscular atrophy (SMA) is one of the most common childhood onset neurodegenerative disorders in global health whereby novel biomarkers and therapeutic targets are sorely needed. SMA is an autosomal recessive genetic disorder resulting in degeneration of α-motor neurons in the brain stem and spinal cord that leads to mortality in infants worldwide. In majority of the patients, SMA is caused by homozygous deletion of the SMN1 gene. The clinical spectrum of the SMA displays, however, large person-to-person variations where the underlying mechanisms are poorly understood. We report in this study transcriptomics insights gleaned from patients with the severe type I (GM03813 and GM09677) and the mild type III. Pathway enrichment and functional analysis showed that especially extracellular matrix (ECM), synapse organization, and ECM receptor interaction pathways were affected. Among the neural ECM components, hyaluronan and proteoglycan link protein (HAPLN1), which is a key triggering molecule of the perineuronal net (PNN), was significantly downregulated in type I fibroblasts compared to type III. PNN is a specialized form of neural ECM around the neuronal cell bodies and dendrites in the central nervous system. In addition, we evaluated the PNN expression in vitro in a model established by SMN silencing in the PC12 rat pheochromocytoma cell line which can be differentiated into neurons with nerve growth factor treatment. In this neuronal in vitro model, we found that HAPLN1 showed a significant 50% decrease. Our results describe the association between PNN elements, especially HAPLN1, and SMA pathophysiology for the first time. These observations collectively inform future translational research on SMA for discovery of novel molecular targets for diagnostics and precision medicine innovation.
Introduction
P
The most important pathway among them is the RhoA/ROCK signaling that controls actin cell cytoskeleton organization cell proliferation, cell adhesion, and cell migration (Wu et al., 2005). In 2010, Carulli and Kwok showed that in the absence of HAPLN1, the structure of PNN was impaired. They also demonstrated that after increasing the expression of aggrecan, HAPLN1 and tenascin in vitro more compact PNN structure was formed (Carulli et al., 2010; Kwok et al., 2010).
In many neurodegenerative diseases, a reduced expression of PNN components associated with a neuroprotective role was reported (Bonneh-Barkay and Wiley, 2009). Although the relationship between PNN elements and pathogenesis of many neurodegenerative diseases has been indicated (Hayashi et al., 2002; Hayashi, 2009), there are no data about the role of these proteins in spinal muscular atrophy (SMA), one of the most common childhood onset neurodegenerative genetic disorders, affecting 1:10,000 live births with substantial mortality.
According to the age of onset and clinical severity, SMA is classified as type I, II, and III. The most severe form, type I (Werdnig–Hoffmann), is characterized by muscle weakness and hypotonia, and patients die before the age of 2 years. In type II, the moderate form, patients have the ability to sit; however, they cannot walk without help. In type III, the mildest form of the disease, the symptoms begin after 18 months. Even though patients have the ability to sit and stand in the early years of disease, they become wheelchair-bound later (Pearn, 1980).
The gene responsible from SMA is “Survival Motor Neuron 1” (SMN1) gene located on chromosome 5q13.2 (Lefebvre et al., 1995). Exons 7 and 8 of the SMN1 gene have homozygous deletions in 98% of all types of SMA patients (Rodrigues et al., 1995). SMA patients with broad phenotypic spectrum have the same SMN1 gene deletion regardless of clinical type. This attracts attention to another gene, SMN2, which is a highly homologous gene of SMN1, present in the same chromosomal location 5q13.2 and is retained in all patients (Coovert et al., 1997). The cytosine/thymine change in the exon 7 alters SMN2 mRNA splicing, leading to the skipping of exon 7 and production of SMNΔ7 protein, which is 90% unstable and is degraded.
In contrast, SMN2 gene produces full-length SMN protein which is present in low amounts (10%) (Lorson et al., 1999). Although this amount is not sufficient for the survival of motor neurons, SMN2 is regarded as a modifier of disease outcome.
The number of copies of SMN2 shows an inverse correlation with disease severity where mild patients (type III) have more copies than severe ones (type I). It has been shown that in type I patients two copies, in type II three copies, and in type III four SMN2 copies are present (Lefebvre et al., 1997; Mailman et al., 2002). However, it is still difficult to explain the mechanism of clinical severity and its correlation with genotype as SMN2 is not the only phenotypic modifier.
So far there is no effective treatment available for SMA. Therefore there is a need to clarify the underlying molecular mechanism of the disease and to determine targets related to the phenotypic variability that can then lead to design and development of novel targeted therapies. With omics technologies and precision medicine approaches emerging in healthcare, accurate phenotypic stratification of patients may be feasible.
In this study, we performed transcriptomic evaluation of fibroblasts of SMA patients with different clinical severity to identify differentially expressed genes and to discover a network involved in SMA pathology. We also established an in vitro disease model by SMN knockdown in neuron like PC12 cell line. The outline of the study design is given in Figure 1. Our findings indicate that PNN may potentially be a novel pathway affected and may provide new insights into molecular pathology of SMA.
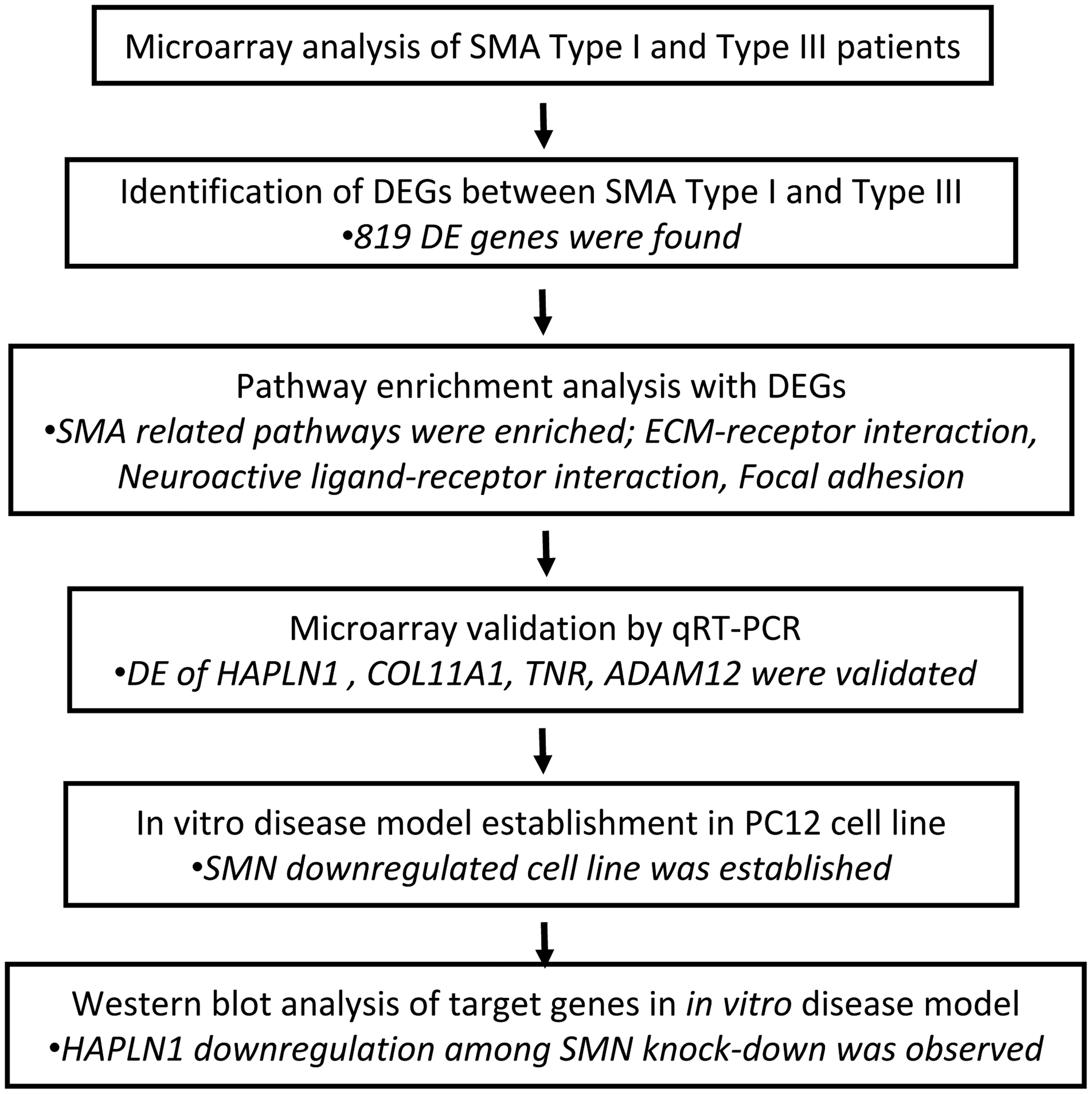
Study design.
Materials and Methods
Transcriptome analysis
Fibroblast cell lines from two SMA type I patients who have SMN1 exon 7 and 8 deletion and two SMN2 copies (one is a 3 year old, GM03813; Coriell Cell Repository and the other one is a 2 year old, GM09677; Coriell Cell Repository) were maintained. In addition, fibroblast culture was established from skin biopsy of a 4-year-old SMA type III patient who have SMN1 exon 7, 8 deletion and three SMN2 copies (Patient No: 1). Cells were incubated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, and 1% L-glutamine (Biochrom, Berlin, Germany) at 37°C with 5% CO2.
Written and informed consent was obtained from the parents of the type III patient. Experimental protocols were approved by the Hacettepe University Faculty of Medicine Ethics Committee (HEK 12/02-13).
The gene expression profiles of type I fibroblasts were compared to type III using GeneChip Human Exon 1.0 ST Array (Affymetrix). Two technical replicates for each sample were used. Microarray data were analyzed in biometric research program (BRB)-Array Tools that is an Excel-integrated tool for the statistical analysis of expression data (Richard et al., 2007). Raw data were normalized using robust multiarray average (RMA) normalization. RMA is one of the most commonly accepted methods for analyzing microarray expression data (Irizarry et al., 2003; Lim et al., 2007). Class comparison tool was used to find out differentially expressed genes [p < 0.05, 1.5-fold change (FC)] between mild and severe groups. The pathway enrichment analysis was performed with the differentially expressed genes using WebGestalt, which is a functional enrichment analysis web tool.
Cell culture
Proliferation of PC12 cells was performed in a complete medium containing high glucose DMEM with stable glutamine, 10% (v/v) horse serum (Biochrom), 5% (v/v) heat inactivated FCS, 1 mM sodium pyruvate, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. The cells were grown in poly-L-lysine hydrobromide (Sigma-Aldrich) coated 75 mm2 tissue culture flasks and incubated at 37°C in a 5% CO2 humidified atmosphere. Cell culture medium was changed every 2 days.
Differentiation of PC12 cell line
Cells were plated into cell culture dishes (1 × 106 cells/dish) for protein isolation. Dishes were coated with poly-L-lysine hydrobromide according to manufacturer's instructions (Sigma-Aldrich). The cells were differentiated for 3 and 5 days in differentiation medium containing high glucose DMEM with stable glutamine, 1% (v/v) horse serum (HS), 1 mM sodium pyruvate, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 50 ng/mL fresh nerve growth factor (NGF) (Sigma-Aldrich).
Transfection
SMN specific siRNA against rat SMN1 gene exon 5 (Ambion; SMN1 Silencer Select Predesigned siRNA-S133927) was transfected with Lipofectamine RNAiMAX (Invitrogen) in six-well plates (3 × 105 cells/well). Three microliters of Lipofectamine was diluted in 50 μL differentiation medium incubated for 5 min. Various siRNA concentrations (50, 60, and 70 pmol) were applied to get maximum transfection and SMN knockdown efficiency. Negative control siRNA 50 pmol (Ambion; Silencer Negative Control No. 2 siRNA) that has no significant sequence similarity to mouse, rat, or human genes was used for determining transfection efficiency and for eliminating the off-target effects.
Following 24 h transfection siRNA mixture was removed and replaced with differentiation medium. After 3 days of differentiation cells were collected for protein isolation. SMN knockdown levels were determined by comparing the cells transfected with negative control siRNA. After 72 h differentiation, siRNA transfection was repeated to obtain further differentiated SMN knockdown PC12 cells. After incubation for 24 h siRNA mixture was removed and replaced with differentiation medium. After differentiation day 5, cells were collected for protein isolation.
Western blot analysis
PC12 cells were scraped and lysed in lysis buffer pH: 7.4 (10 mM Tris-base, 300 mM NaCl, 2 mM EDTA, and 0.5% Triton-X-100 with one mini protease inhibitor tablet). Samples were then sonicated on ice and centrifuged at 17,000 g for 10 min at 4°C. Protein concentrations were determined using the bicinchoninic acid assay and stored at −80°C until use. Proteins (50 μg) were denatured with 4 × Laemmli buffer and boiled at 100°C for 5 min. Samples were loaded on 8–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then blotted onto nitrocellulose membranes. Eight percent SDS-PAGE gel and wet transfer method (30 V, overnight) were used for imaging TNR and ACAN. Twelve percent SDS-PAGE gel and semidry transfer method (25 V, 30 min) were used for imaging SMN and HAPLN1.
Membranes were blocked with 10% non-fat dry milk in Tris-buffered saline with 0.2% Tween-20 (0.2% TBS-T) for 1 h and then incubated with following primary antibodies overnight at 4°C: Mouse anti-SMN (1:1000; BD Transduction Laboratories 610646), goat anti-HAPLN1 (1:1000; R&D Systems AF2608), rabbit anti-ACAN (1:500; Merck AB1031), mouse anti-TNR (clone #619 MAB1624, 1:500; R&D Systems Oxfordshire, UK), and mouse anti-GAPDH (1:5000; Sigma). Membranes were then incubated with horseradish peroxidase-coupled secondary antibodies at a dilution of 1:1000. They were rinsed thrice in 0.2% TBS-T, and signals were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Statistical analysis
The western blot bands of SMN knockdown and control cells were measured using Gel Analysis function of ImageJ software. The alterations in protein level were statistically analyzed with the nonparametric Kruskal–Wallis test, using the GraphPad Prism (6.01) software. Values of p < 0.05 were considered significant. Graphics were drawn with mean values using the GraphPad Prism (6.01) software with error bars equivalent to the standard error of the mean.
Results
Microarray analysis of expressed genes in severe and mild SMA patients
The microarray analysis was performed with severe type I SMA fibroblast cell lines (GM03813 and GM09677) and mild type III SMA fibroblast cell line (Patient No: 1). Eight hundred nineteen genes were found to be differentially expressed between these groups (p < 0.05, Supplementary Table S1). Among the differentially expressed genes, 323 were found to be upregulated while 496 were found to be downregulated.
Among these transcripts, HAPLN1 was observed to be significantly downregulated 6.26-folds in severe case (p = 0.024). This transcript was related to ECM-neuron interaction and was one of the main components of PNN formation. Also detecting ACAN, TNR, COL11A1 (p = 1.46E-05), and ADAM12 (p = 0.005) transcripts has attracted attention because of their role in synapse formation, regulation, and signal transduction between neuron and ECM. While the changes in the expression were not significant in ACAN and TNR among the two different groups, as these transcripts were part of PNN components and had crucial role in neurodegeneration, they were selected as targets (Table 1).
FC, fold change.
Pathway enrichment analysis
Among the KEGG pathways that were enriched in WebGestalt analysis, the ECM-receptor interaction (p = 0.0007), neuroactive ligand-receptor interaction (p = 0.001), and focal adhesion (p = 0.0014) were the prominent ones, which could be important in SMA because of their potential roles in synaptic stability, regulation of plasticity, signal transduction through a neuron, and neuroprotective role of ECM-neuron interaction (Table 2).
ECM, extracellular matrix.
qRT-PCR results validated the microarray data
The qRT-PCR performed for the genes taking part in PNN structure and ECM-receptor interaction pathway validated the microarray results. HAPLN1, which is an essential structural element of PNN, was found to be threefold downregulated in qRT-PCR experiments concordantly with microarray results. COL11A1, which is one of the genes taking part in ECM-receptor interaction pathway, was also found to be downregulated 10.6-folds in severe SMA patient compared to mild case also validating the array result. In addition TNR and ADAM12 were found to be downregulated in fibroblasts of severe SMA patients compared to that of mild one (Fig. 2).

Genes related to PNN and ECM were downregulated in severe patients compared to mild. ECM, extracellular matrix; PNN, perineuronal net.
In vitro disease model was established by knockdown of SMN in PC12 cells
PC12 cells were differentiated, and at day 3, maximum SMN downregulation, which was 75%, was obtained with 50 pmol siRNA transfection (Fig. 3A, B). Knockdown efficiency was decreased to 50% when the cells were transfected with 60 and 70 pmol siRNA. At differentiation day 5, due to lack of proliferation in terminally differentiated cells, SMN knockdown efficiency was drastically reduced to 20% (Fig. 3C, D). The differentiation experiments were no longer carried out to day 7. Therefore an in vitro SMA model was established by RNA interference technology, and for further analysis, the study was carried out using 50 pmol SMN siRNA transfection.
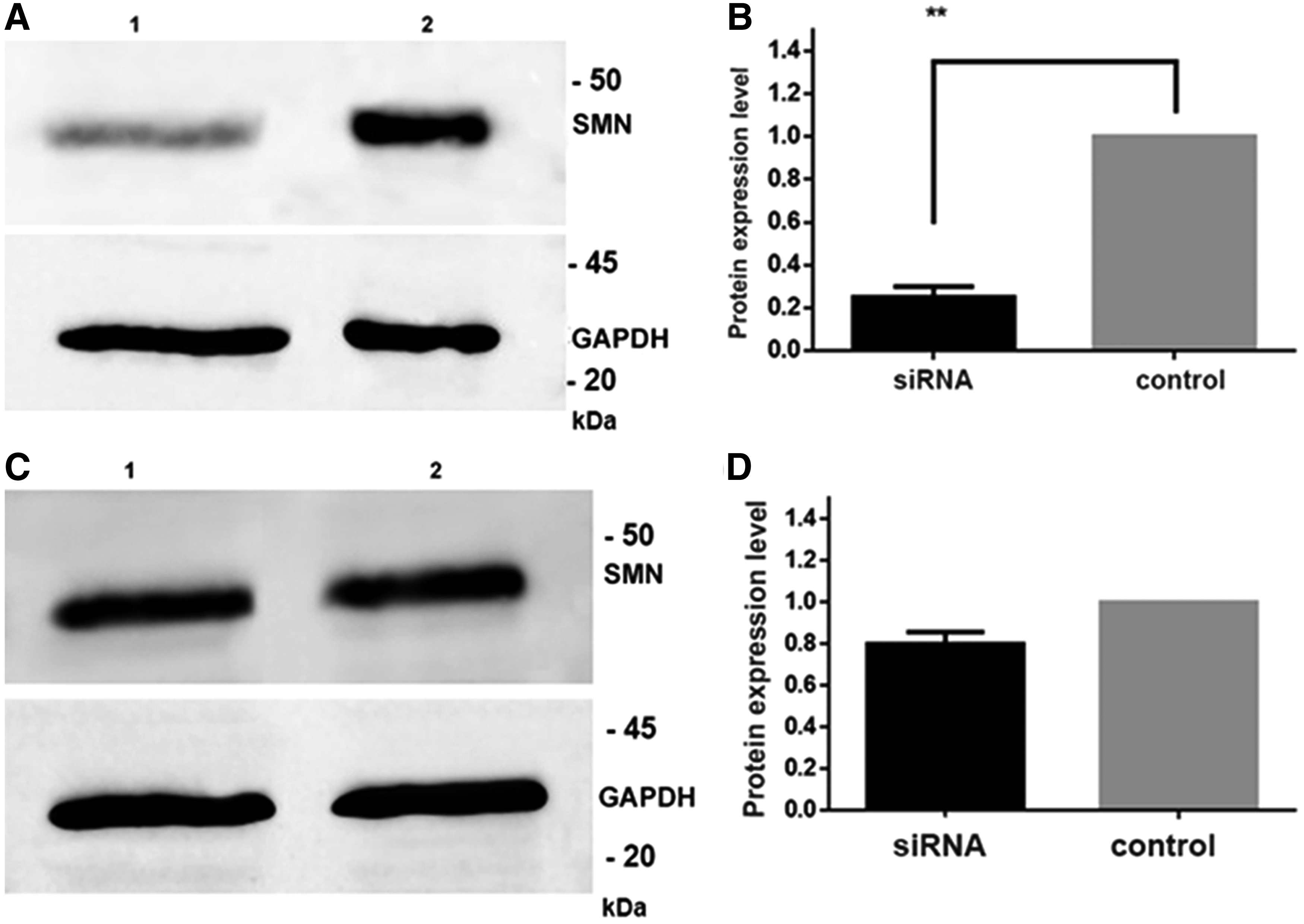
Representative western blot analysis of SMN protein and protein expression graphic in SMN knockdown (lane 1) and control (lane 2) PC12 cells differentiated for 3 days (
Altered expression of PNN elements in SMN depleted cells
In this in vitro SMA model, the expression of HAPLN1, ACAN, and TNR was investigated at the protein level. HAPLN1 protein levels showed 50% decrease in SMN depleted cells (Fig. 4A, B). However, TNR and ACAN protein levels did not show any change compared to negative control siRNA (p < 0.05) (Fig. 5A, B).
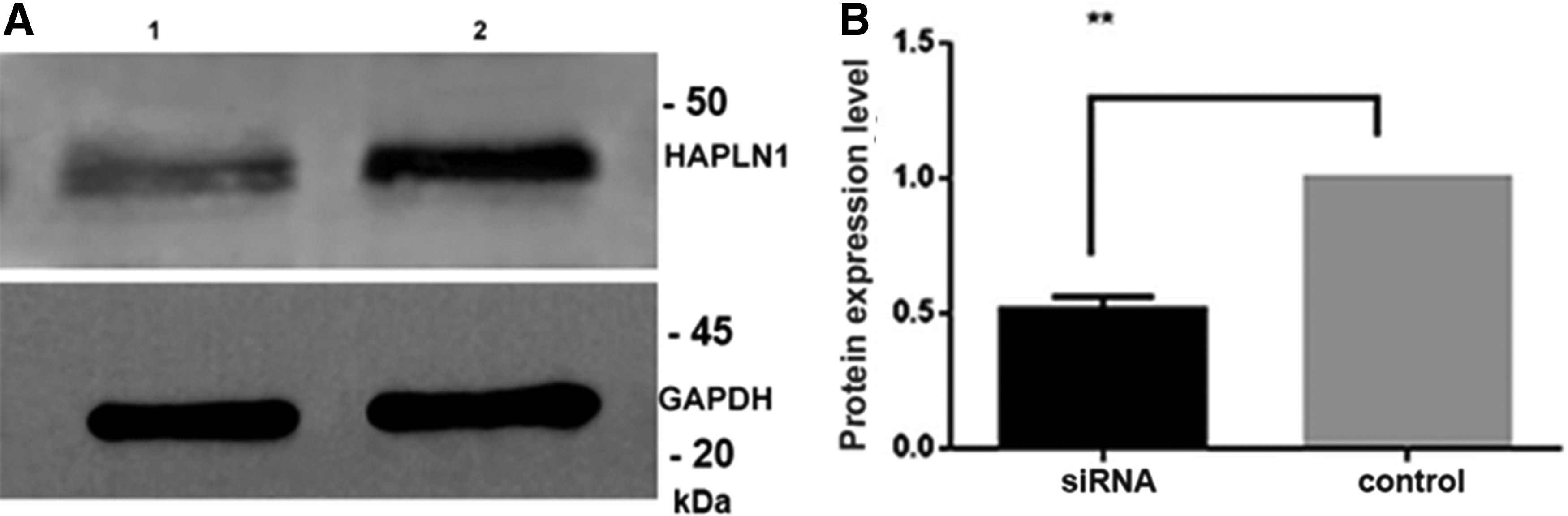
Representative western blot analysis
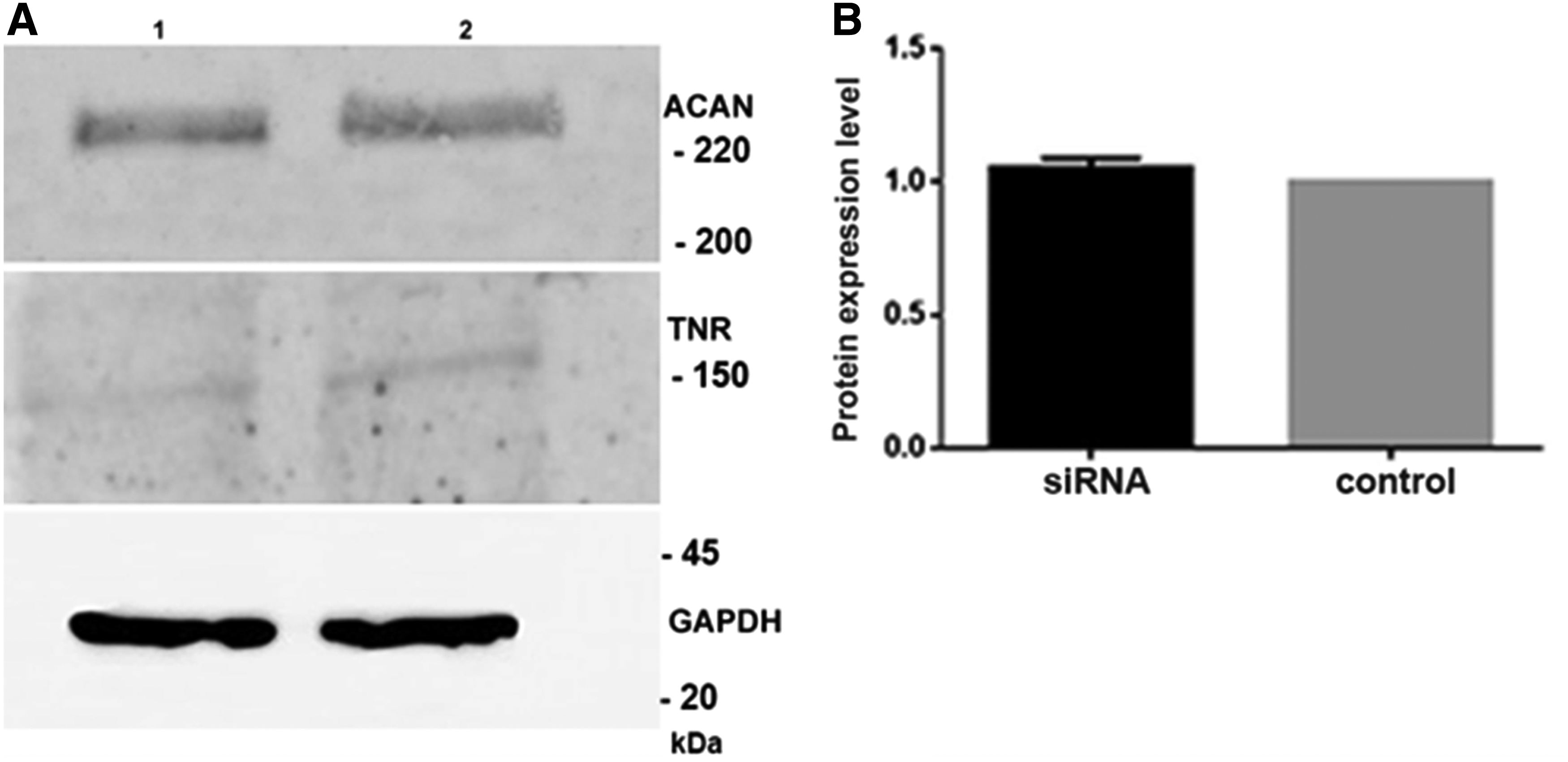
Representative western blot analysis of TNR, ACAN proteins
Discussion
A highly organized structure that surrounds neurons, known as PNN, plays role in stabilization of synaptic interactions, regulation of synaptic plasticity, and protection of neurons from oxidative damage by neutralizing the oxidative potential in neuronal microenvironment (Fiedler et al., 2007; Suttkus et al., 2014). Recent studies have shown that in neurodegenerative diseases like Alzheimer's, multiple sclerosis, and ischemic stroke, the structural integrity of PNN elements was found to be disrupted, and neuronal function and survival was found to be decreased (Bonneh-Barkay and Wiley, 2009; Lemarchant et al., 2017; Sethi and Zaia, 2017; Suttkus et al., 2016).
In another study on Alzheimer's patients, it was shown that cortical areas poor of PNN were prone to neurofibrillary degeneration much more than others (Morawski et al., 2004). In epilepsy models, aggrecan and HAPLN1 RNA/protein expressions were decreased and PNN structure was impaired (McRae and Porter, 2012; McRae et al., 2012). Also in amyotrophic lateral sclerosis which is a motor neuron disease, when cerebrospinal fluids of patients were collected and compared to postmortem control samples, tenascin expression was decreased and morphology of PNN was disrupted (Forostyak et al., 2014).
So far there are no data available about PNN in SMA. The SMN1, the key gene affected in SMA, is deleted in patients but the SMN2 gene located at the same region, which shows high homology to SMN1, is retained (Coovert et al., 1997). SMN2 copy number influences clinical severity.
In recent years, there has been an effort to search for modifier genes in SMA families with affected siblings that show variable clinical severity. Studies on modifier genes such as plastin 3 (PLS3) and neuritin 1 (NRN1) genes have conflicting results, suggesting that novel genetic modifiers should be identified and characterized (Bernal et al., 2011; Oprea et al., 2008; Stratigopoulos et al., 2010; Yener et al., 2017).
SMA is described as a multisystem disorder because due to SMN protein deficiency, various cellular pathways such as RNA metabolism, actin dynamics, vesicular transport, and endocytosis are affected (Crawford and Pardo, 1996). The molecular pathogenesis of the disease and underlying mechanism is still not known. Although type I and type III SMA patients have the same deletions in SMN1 gene, they show different clinical features and severity. This phenotypic variability makes genotype–phenotype correlation difficult to establish.
Fibroblasts are the cells that show expression of the genes associated with neurological diseases and also reflect brain transcriptome. Due to inaccessibility of the target tissue such as motor neurons, fibroblasts can be used as a good source for disease modeling in neurology (Visser et al., 2010). We performed transcriptomic analysis and compared the fibroblasts obtained from severe type I and mild III patients. As type II is the phenotypically intermediate form, we focused on the transcriptomic differences between these two clinically extreme groups to unveil the cause of severity.
Our results showed that differentially expressed genes between severe/mild groups and significantly affected pathways are the ones that have a role in ECM elements, synapsis organization, and ECM-receptor interaction. Detailed analysis on pathways and functional-related gene groups showed that especially HAPLN1, ADAM12, and COL11A1 were downregulated in type I fibroblasts compared to type III. These results pointed out that alteration and disorganization of the PNN architecture may be associated with SMA severity. Although the sample numbers were low that were representing the two cases (severe vs. mild), the strong relations of the differentially expressed genes with the molecular pathways that related to SMA phenotype may show the robustness of the microarray data obtained even from limited number of samples.
It is known that HAPLN1, which is a link protein, is one of the essential components of neural ECM and initiates formation of PNN network (Carulli et al., 2010; Horii-Hayashi et al., 2017). The other component, TNR, is responsible for strong binding of the PNN elements to each other (Chiquet-Ehrismann, 2004; Mouw et al., 2014). ACAN, which is a member of lectican, has a role in maintenance of an intact and complex PNN structure (Giamanco et al., 2010; Miyata and Kitagawa, 2016). Although the change in the expressions of TNR and ACAN was not significant in our study, it was reported by Galtrey et al. (2008) that they were essential elements for PNN formation while other elements were optional. Based on these data, we analyzed the essential molecules of PNN in which HAPLN1 plays a fundamental role.
To investigate PNN expression in a cell type that is more relevant to the disease, we established SMN gene silenced in vitro model using neuronal PC12 cells. With NGF, these cells can be differentiated into neurons homogenously and produce neurites which is one of the advantages of PC12 cells. Several studies described different techniques on PC12 transfection and SMN knockdown efficiency, mostly about stable transfection with shRNA at 3-day differentiated cells (Hensel et al., 2014; Mattis et al., 2008; Nölle et al., 2011; Van Bergeijk et al., 2007; Zou et al., 2011).
Among all articles, transient transfection with siRNA after 3 day differentiation showed a 70–75% knockdown of SMN protein which was similar to our results. Also Bowerman et al. (2007) showed that after 7 day differentiation cells were not fully differentiated, and the number of differentiated cells was significantly decreased compared to wild-type cells. In our experiments we also did not extend differentiation period to 7 days. Our data demonstrated that at differentiation day 5, SMN knockdown efficiency was reduced to 20%.
In this SMN knockdown model, we analyzed the expression of HAPLN1, ACAN, and TNR. We found that HAPLN1 protein levels were decreased but TNR and ACAN levels remained unchanged. These data support our hypothesis that main components of PNN, especially HAPLN1, were affected in an in vitro model of SMA.
Among PNN components, HAPLN1 has a pivotal role. It enhances the interactions of chondroitin sulfate proteoglycans with hyaluronan. These components determine the scaffold of PNN, and their interaction increases the stability of PNN structure (Dzyubenko et al., 2016). It is also known that HAPLN1 is the major element that triggers PNN meshwork formation, and its expression changes due to disease or healthy state. We observed 50% decrease in HAPLN1 expression level in SMN knockdown PC12 cells compared to negative control siRNA transfected cells.
ACAN is one of the essential chondroitin sulfate proteoglycans in the PNN, and in the lack of ACAN, PNN structure cannot be established properly. Also it is shown that in ACAN knockout mouse models, ACAN is required for survival (Giamanco et al., 2010; Miyata and Kitagawa, 2016). We found that in SMN knockdown cells ACAN levels did not decrease, they remained unchanged. These data suggest that SMN deficient cells that have insufficient amount of HAPLN1 may need ACAN for PNN formation and survival.
Although the exact role of TNR is not known, PNN structure is mediated by TNR. As it functions as a trimeric glycoprotein, TNR has the ability to cross-link PNN components due to its structure. As reported by Giamanco and Matthews, (2012) TNR is the final element that is recruited to form the PNN structure. Also it undergoes several posttranslational modifications, which makes it more stable. In our previous study performed on wild-type differentiated PC12 cells, we found that TNR levels did not show any change at differentiated cells (day 3) compared to nondifferentiated state (Eskici et al., 2018). In this study, TNR level showed very low expression that could not be quantitated both in SMN knockdown and negative control siRNA treated PC12 cells. Our data indicate that at differentiation day 3, TNR may not have participated into the PNN composition yet.
In recent years glycomics has attracted attention in its application in precision medicine (Wang et al., 2016). Glycans were found to be involved in some inflammatory and autoimmune diseases. They can also take part in pathways triggering inflammation. Different glycosylation profiles were detected in complex diseases such as ischemic stroke and Parkinson's disease compared to controls (Liu et al., 2018; Russell et al., 2017). These findings indicate that glycans can also be developed as diagnostic, prognostic, and therapeutic biomarkers (Russell et al., 2018). As HAPLN1, which interacts with chondroitin sulfate proteoglycans and allows the participation of other PNN elements, is associated with type I fibroblasts compared to type III, it may be regarded as a biomarker for SMA in the future.
In this study, we tested our hypothesis that alteration of PNN elements may be associated with pathophysiology of SMA. The assessment of PNN function in SMA pathophysiology has not been explored previously. Among the molecules that were essential for PNN formation, we found that only HAPLN1, which plays a fundamental role, was downregulated in SMA model. Our study provided new insights into ECM and neuron interaction pathways in neurodegeneration. In addition, it revealed involvement of PNN, especially HAPLN1, as the reason for phenotypic variability among SMA patients with the same genotype.
For future studies, detecting impaired PNN structure and loss also in SMA mouse models will provide opportunity for identification of drug candidate molecules that target PNN. Therapeutic treatments directed to maintain or restructure the integrity of PNN may represent a novel strategy not only in SMA but also in other neurodegenerative diseases in which PNN is affected. Deciphering the underlying mechanisms of SMA will pave the way toward phenotypic characterization and new targeted and personalized therapies.
Footnotes
Acknowledgments
This research was supported by The Scientific and Technological Research Council of Turkey, TUBITAK, Project no: 114S914. Microarray consumables and reagents were purchased from a part of overhead (indirect costs) of TUBITAK, Project no: 105G014.
Authors' Contributions
All authors contributed to revisions for significant intellectual content and approved the final version of the article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.