
Abstract
Abstract
Nontuberculous mycobacterial (NTM) species present a major challenge for global health with serious clinical manifestations ranging from pulmonary to skin infections. Multiomics research and its applications toward clinical microbial proteogenomics offer veritable potentials in this context. For example, the Mycobacterium abscessus, a highly pathogenic NTM, causes bronchopulmonary infection and chronic pulmonary disease. The rough variant of the M. abscessus UC22 strain is extremely virulent and causes lung upper lobe fibrocavitary disease. Although several whole-genome next-generation sequencing studies have characterized the genes in the smooth variant of M. abscessus, a reference genome sequence for the rough variant was generated only recently and calls for further clinical applications. We carried out whole-genome sequencing and proteomic analysis for a clinical isolate of M. abscessus UC22 strain obtained from a pulmonary tuberculosis patient. We identified 5506 single-nucleotide variations (SNVs), 63 insertions, and 76 deletions compared with the reference genome. Using a high-resolution LC-MS/MS-based approach (liquid chromatography tandem mass spectrometry), we obtained protein coding evidence for 3601 proteins, representing 71% of the total predicted genes in this genome. Application of proteogenomic approach further revealed seven novel protein-coding genes and enabled refinement of six computationally derived gene models. We also identified 30 variant peptides corresponding to 16 SNVs known to be associated with drug resistance. These new observations offer promise for clinical applications of microbial proteogenomics and next-generation sequencing, and provide a resource for future global health applications for NTM species.
Introduction
N
Studies have shown that M. abscessus has two major morphotype forms, rough (R) and smooth (S) (Byrd and Lyons, 1999; Ripoll et al., 2009). The rough (R) form is known not to have surface polyketide compound, glycopeptidolipids, and is associated with more severe infection (Catherinot et al., 2007; Howard et al., 2006; Pawlik et al., 2013). M. abscessus UC22 strain has the rough (R) morphological form and is one of the causative organisms of the upper lobe fibrocavitary (UC) type of pulmonary disease (Kim et al., 2017).
Virulence in mycobacteria is related to the ability of the bacterial cells to survive in the host (Smith, 2003). In mycobacterial genomes, most of these virulence genes encode enzymes for lipid and fatty acid metabolism, cell envelope proteins, gene expression regulators, and proteins of signal transduction systems (Forrellad et al., 2013). Virulence factors such as PE/PPE family, mammalian cell entry (Mce) protein family, ESX (type VII secretion systems) export, and twin-arginine translocase (Tat) export systems have been well characterized in Mycobacterium tuberculosis but have not been studied in M. abscessus (Bottai and Brosch, 2009; Bottai et al., 2014; Tsirigotaki et al., 2017; Zhang and Xie, 2011).
A recently published study has shown that rough morphotype of M. abscessus is generally more virulent than the smooth morphotype, which suggests interdependence of morphology and virulence (Kim et al., 2017). M. abscessus UC22 strain has a rough morphotype and there have been no studies on integrated molecular profiling of M. abscessus UC22 strain so far, for the identification of genomic determinants of virulence and drug resistance, which may be associated in the UC form of pulmonary disease.
In addition, the clinical features presented in pulmonary tuberculosis infection, as well as in the pulmonary infection due to M. abscessus, are reported to be similar (Ishiekwene et al., 2017; Wankhade and Bhore, 2017). As a result, a clear distinction between them is currently difficult. Furthermore, M. abscessus is known to be inherently resistant to several chemotherapeutic agents used to treat pulmonary tuberculosis (Griffith, 2014). Therefore, lack of early detection results in inappropriate treatment of M. abscessus infection. This necessitates a need for a detailed molecular profiling of M. abscessus that will provide a platform to further investigate differentiating molecular patterns.
The advancement in high-throughput mass spectrometry has not only enabled the in-depth exploration of proteomes but also helped in refining the gene models of several organisms from bacteria to human (Gallien et al., 2009; Heunis et al., 2017; Kim et al., 2014; Venter et al., 2011). It is reasonable to believe that protein-level evidence as provided by LC-MS/MS analysis is more confirmatory than the computational prediction of protein-coding genes annotated against any genome sequence (Ruggles et al., 2017). The multidisciplinary protocols of integrating proteomic data into the genome annotation process are collectively termed proteogenomics (Venter et al., 2011).
Several proteogenomic studies on mycobacterium species have highlighted the limitations of gene prediction programs, including genome assembly errors, start site, and gene boundaries (Gallien et al., 2009; Kelkar et al., 2011; Pinto et al., 2018; Potgieter et al., 2016; Zheng et al., 2017).
In this study, we carried out next-generation sequencing and LC-MS/MS analysis of M. abscessus UC22 clinical isolate, and applied a proteogenomic approach to improve the annotation of the genome. We identified several single-nucleotide variations (SNVs) in the genes known to be associated with drug resistance, validated further by the identification of the corresponding variant peptides through the proteomic data. Use of a proteogenomic approach in the current study led to the discovery of novel protein-coding regions in the M. abscessus UC22 genome, which have been missed in the previous annotation. We believe that the integrated multiomic approach presented in this study enhanced our knowledge on the M. abscessus UC22 strain. The results of which can provide insight into understanding the molecular mechanisms of virulence and drug resistance in this strain.
Materials and Methods
M. abscessus culture and whole-genome sequencing
A clinical isolate from pulmonary tuberculosis patient (Patient ID: JAL-14421) was obtained from the Department of Microbiology, National JALMA Institute of Leprosy and other Mycobacterial Diseases, Agra, India. The sputum was smear positive for acid-fast bacilli. Culturing and biochemical tests revealed the isolate to be an NTM. The drug susceptibility testing revealed the isolate to be resistant to rifampicin, isoniazid, ethambutol, and streptomycin. The isolate was processed as per the standard protocol and inoculated on Lowenstein-Jensen (LJ) slants and then was further used for DNA extraction. The DNA isolation was carried out similar to Sharma et al. (2017). We used the cetyltrimethylammonium bromide method of DNA isolation from the bacterial culture grown on LJ slant (van Embden et al., 1993).
The quality of DNA was assessed on 1% agarose gel and the DNA was quantified on the NanoDrop spectrophotometer (ND-1000 UV-Vis). Paired-end DNA libraries were prepared according to the manufacturer's description and were sequenced with a read length of 2 × 100 nucleotides using an Illumina HiSeq2500 instrument (Illumina, Inc., San Diego). Whole-genome sequencing led to the identification of the isolate to be M. abscessus. Metagenomic analysis was performed using Kraken tool on the raw reads, which confirmed the isolate belonged to M. abscessus UC22 strain (Wood and Salzberg, 2014). The study was approved by the National JALMA Institute of Leprosy and other Mycobacterial Diseases Institutional Ethics Committee.
M. abscessus culture and protein extraction
M. abscessus UC22 clinical isolate was grown in Middlebrook 7H9 medium (supplemented with OADC [oleic acid, dextrose, and catalase]). The cultures were incubated at 37°C in shaker incubator. The culture was harvested at stationary phase and centrifuged for proteomic analysis. The cell pellets were washed thrice with chilled phosphate-buffered saline. Mechanical lysis was carried out with 0.1 mm Zirconia beads in a Precellys tissue homogenizer for 3 min (6500 rpm) twice under continuous cooling at or below 4°C in 2% SDS lysis buffer (2% SDS in 50 mM triethylammonium bicarbonate (TEABC), 1 mM sodium fluoride, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerophosphate).
Lysates were resuspended in twice the volume of lysis buffer with phosphatase inhibitors, and the lysates were passed through 0.22 μm filters. Bicinchoninic acid assay was used to estimate the concentration of proteins.
Trypsin digestion and fractionation
In-solution digestion was carried out as described previously (Nagarajha Selvan et al., 2014). Briefly, 300 μg of protein was reduced and alkylated with 10 mM dithiothreitol and 20 mM iodoacetamide, respectively. The proteins were precipitated using chilled acetone kept at −20°C overnight. The protein pellets were reconstituted in 100 mM TEABC buffer and digested using trypsin (1:20) (modified sequencing grade; Promega, Madison, WI) at 37°C overnight. After confirming the digestion efficiency, 100 μg peptide digest was fractionated using StageTip-based strong cation-exchange (SCX) chromatography.
The remaining peptide digest was subjected to basic pH reverse-phase chromatography (bRPLC). For bRPLC fractionation, the peptide digest was loaded onto Waters XBridge column (Waters Corporation, Milford, MA; 130 Å, 5 μm, 250 × 4.6 mm) using a manual injector attached to the Hitachi HiChrom HPLC system. The peptide digest was fractionated at a flow rate of 0.5 mL/min using a linear gradient of 5–100% of 10 mM TEABC, 90% acetonitrile over 120 min. A total of 96 fractions were collected and concatenated to a final of 12 fractions. The pooled fractions were evaporated to dryness using SpeedVac concentrator (Thermo Scientific) followed by desalting using C18 StageTips.
For SCX chromatography-based fractionation, the peptide digest was evaporated to dryness and reconstituted in 2% trifluoroacetic acid (TFA) solution. The sample was loaded onto StageTip packed with cation-exchange material PolySULFOETHYL A (EmporeTM Cation 47 mm extraction disc 66889-U), washed with 0.2% TFA, and fractionated into 6 fractions using increasing concentrations of ammonium acetate (50–300 mM ammonium acetate, 20% acetonitrile, and 0.5% formic acid). The final fraction was obtained by eluting the peptides using 5% ammonium hydroxide and 80% acetonitrile. The eluates were evaporated to dryness using SpeedVac concentrator (Thermo Scientific) at room temperature and stored at −20°C until LC-MS/MS analysis.
LC-MS/MS analysis
The peptides from both bRPLC (12 fractions) and SCX (6 fractions) fractionation methods were analyzed in technical replicates. Two separate fractionation methods were used to increase proteome coverage. The data were acquired on Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer (Thermo Fischer Scientific, Bremen, Germany) connected to Easy-nLC-1200 nanoflow liquid chromatography system (Thermo Scientific). The peptides were reconstituted in 0.1% formic acid and loaded onto a 2 cm trap column (nanoViper, 3 μm C18 Aq) (Thermo Fisher Scientific). Peptides were separated using a 15 cm analytical column (nanoViper, 75 μm silica capillary, 2 μm C18 Aq) at a flow rate of 300 nL/min. The solvent gradients were set as linear gradient of 5–35% solvent B (80% acetonitrile in 0.1% formic acid) for 90 min. The total run time for each fraction was 120 min.
Global MS survey scan was carried out at a scan range of 400–1600 m/z (120,000 mass resolution at 200 m/z) in a data-dependent mode using an Orbitrap mass analyzer. The maximum injection time was 5 msec. Only peptides with charge state 2–6 were considered for analysis, and the dynamic exclusion was set to 30 sec. For MS/MS (tandem mass spectrometry) analysis, data were acquired at top speed mode with 3-sec cycles and subjected to higher collision energy dissociation with 34% normalized collision energy. MS/MS scans were carried out at a range of 100–1600 m/z using Orbitrap mass analyzer at a resolution of 30,000 at 200 m/z. Maximum injection time was 120 msec. Internal calibration was carried out using lock mass option (m/z 445.1200025) from ambient air.
Data analysis
Mapping and variant detection using M. abscessus UC22 reference genome
The quality of the raw reads were checked for PHRED score of <Q20 using FastQC toolkit. Reads were then aligned to M. abscessus UC22 reference genome (NZ_CP012044.1) using Burrows–Wheeler Alignment Tool (BWA version-0.7.15) (Li and Durbin, 2009). The alignment files were subjected to local realignment and deduplication using the Genome Analysis Toolkit (GATK version-3.6) (McKenna et al., 2010). Variants (SNVs and insertion/deletions (In/Dels) were identified from each alignment file using GATK. The variants and In/Dels were annotated using in-house Perl scripts. Variants identified in this study were manually inspected using Integrative Genomics Viewer (Robinson et al., 2011).
Multilocus sequence typing analysis
To confirm the strain, we carried out multilocus sequence typing (MLST) on M. abscessus UC22 isolate using MLST 1.8 tool (Larsen et al., 2012).
Database searches for peptide and protein identification
Raw data files were processed to generate peak list files using Proteome Discoverer software version 2.1 (Thermo Fisher Scientific, Bremen, Germany). The MS/MS spectra were searched using SequestHT and Mascot search algorithms against protein database (NCBI updated March 2017), combined with common contaminants (total no. of sequences: 5,211). The genome sequence of M. abscessus UC22 was downloaded from the NCBI FTP site (NCBI Reference Sequence: NZ_CP012044.1). A six-frame translated database was created containing M. abscessus UC22 translated sequences from stop codon to stop codon using in-house Python script. Studies have shown that apart from AUG, mycobacteria use GTG and TTG as initiator methionine codons (Cole et al., 1998; Kelkar et al., 2011).
An alternate start codon database was additionally created using GTG and TTG codons and it was translated as initiator methionine in addition to valine and leucine, respectively. Peptide sequences smaller than seven amino acids were not included in the database. The hypothetical N-terminal tryptic peptide database was created by fetching all the peptide sequences that began with methionine and ended with lysine/arginine from the six-frame translated genome database. Peptide sequences with lengths ranging from 7 to 25 amino acids were considered. The raw data files were first searched against M. abscessus UC22 protein database from NCBI (updated March 2017), combined with common contaminants (total no. of sequences: 5,211).
Following the first-pass search, other searches were performed sequentially in the following order (i) against M. abscessus UC22 protein database, (ii) six-frame translated genome database, (iii) alternate start codon database, and (iv) a hypothetical N-terminal peptide database. The search parameters included trypsin as the proteolytic enzyme with a maximum of two missed cleavage allowed. Oxidation of methionine and N-terminal acetylation protein was set as dynamic modifications, whereas carbamidomethylation of cysteine was set as static modification. Precursor ion mass tolerance and fragment ion mass tolerance were allowed with 10 ppm and 0.05 Da, respectively. The false discovery rate (FDR) was calculated using Percolator node and a cutoff of 1% peptide spectral match (PSM) and 1% peptide-level FDR was used for identifications.
Bioinformatic analysis
We carried out in silico analysis with SignalP (V4.1) to identify signal peptides in M. abscessus UC22 proteins (Petersen et al., 2011). Furthermore, the subcellular localization of all the proteins identified in our study was predicted using the PSORTb (V3.0.2) (Yu et al., 2010). We used PRED-LIPO prediction tool to identify lipoproteins in our proteomic data from M. abscessus UC22 clinical isolate (Bagos et al., 2008). To characterize the variant proteins, gene ontology (GO) analysis was performed using Blast2GO tool (Gotz et al., 2008). Functional enrichment of the proteins identified with variant peptides was carried out using FunRich with the Blast2GO annotation as backend database (Pathan et al., 2015).
Generation of customized strain-specific proteome database
A custom variant protein database was created to identify variant peptides containing the nonsynonymous SNVs identified in the genome analysis. Only the nonsynonymous SNVs were incorporated into the protein sequences. The variant peptides having single PSM were manually verified by checking their spectra.
The unmapped reads that did not align to the M. abscessus UC22 reference genome in the genome analysis were assembled with Velvet de novo assembler (Version 1.2.10) (Zerbino and Birney, 2008). VelvetOptimiser (Version 2.2.6) was used to determine the optimal kmer length for de novo assembly of the unmapped reads and was run with default parameters according to the software documentation (Zerbino and Birney, 2008). A kmer length of 75 was used for generation of contigs, further translated in six frames using in-house Python script. The raw data files were searched against the variant protein database and de novo assembled contig database separately with same parameters used in the protein searches.
Workflow for manual genome annotation
The genome search-specific peptides (GSSPs) obtained from searching the MS/MS data against alternate genome translated database were mapped to the M. abscessus UC22 genome to identify the probable coding regions. Genome coordinates of all the peptides were obtained using in-house Perl script. Peptides mapping to multiple locations in the genome were not considered for further analysis.
From the translated genome database search results, GSSPs were obtained by filtering peptides that mapped to known proteins. FgeneSB and GeneMark (Version 2.5) gene prediction programs were used to generate alternative gene models (Besemer et al., 2001). The novel genes and alterative gene models obtained using peptide evidence and gene prediction programs were checked for their conservation across ortholog evidence in Mycobacterium sp. using protein Basic Local Alignment Search Tool (BLAST) algorithm. MS/MS spectra of all the peptides providing evidence of novel genes or changes in gene structure were manually verified.
Analysis of virulence genes
All proteins identified in the proteomic data and genes carrying SNVs in the genomic data were subjected to BLAST analysis against the virulence factor database (Chen et al., 2012). The genes with at least 96% identity and at least 80% sequence coverage that were orthologous to virulence genes were further considered (Choo et al., 2014).
Data availability
Whole-genome sequencing data were deposited in the NCBI SRA database with accession SRP136682. The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD009405.
Results and Discussion
Strain information and genomic characterization of M. abscessus UC22 isolate
M. abscessus UC22 genome is composed of 5.2 million base pairs with 64% GC, guanine-cytosine content encoding for 5104 genes. We identified a total of 5506 nonsynonymous SNVs (Fig. 1) (Supplementary Table S1A) corresponding to 2080 protein coding genes. In addition, we also identified 63 insertions and 76 deletions in M. abscessus UC22 clinical isolate (Fig. 2A) (Supplementary Tables S1B and S1C).
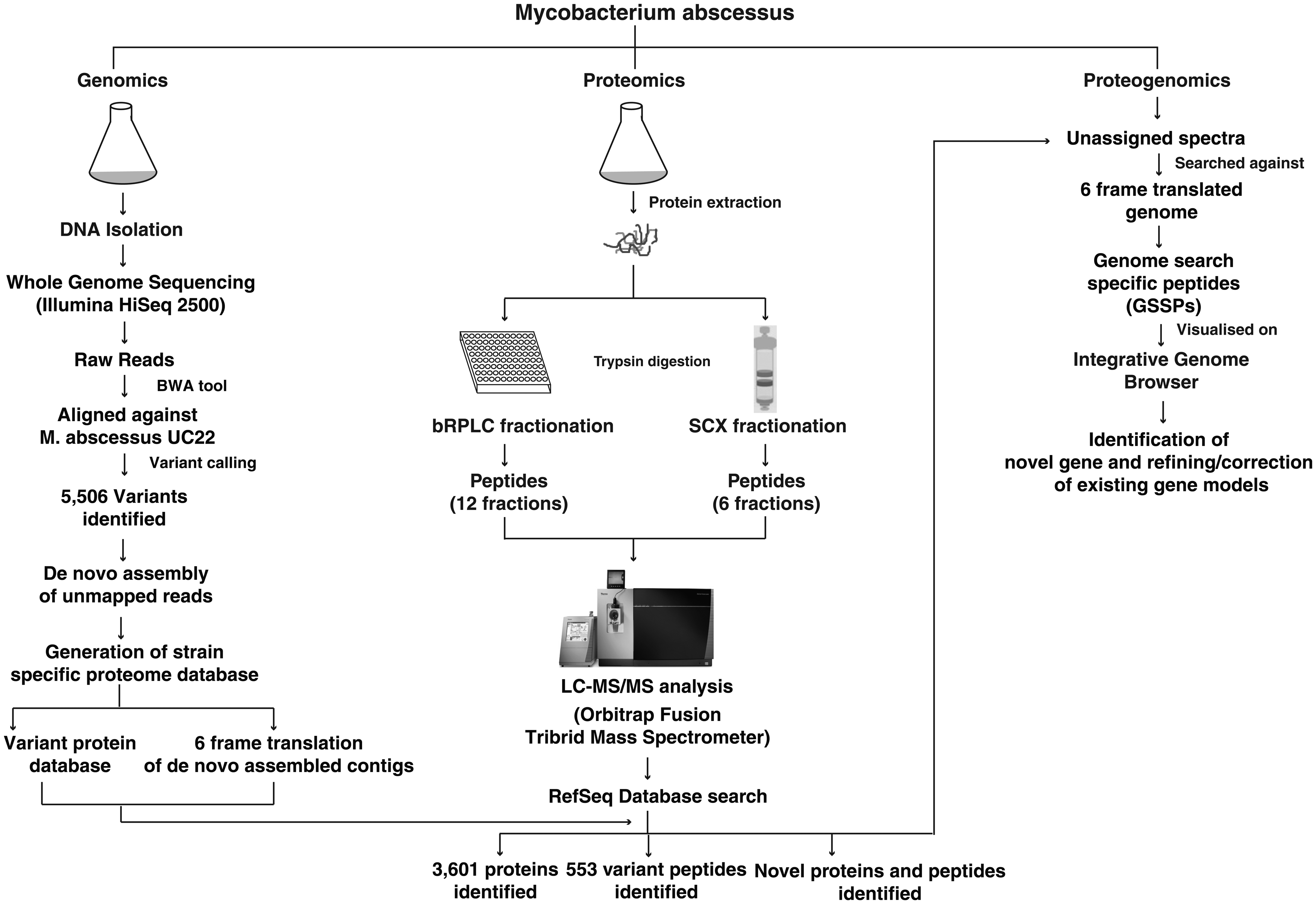
Integrated experimental workflow used to analyze the genome, proteome, and proteogenomics of Mycobacterium abscessus UC22 isolate.
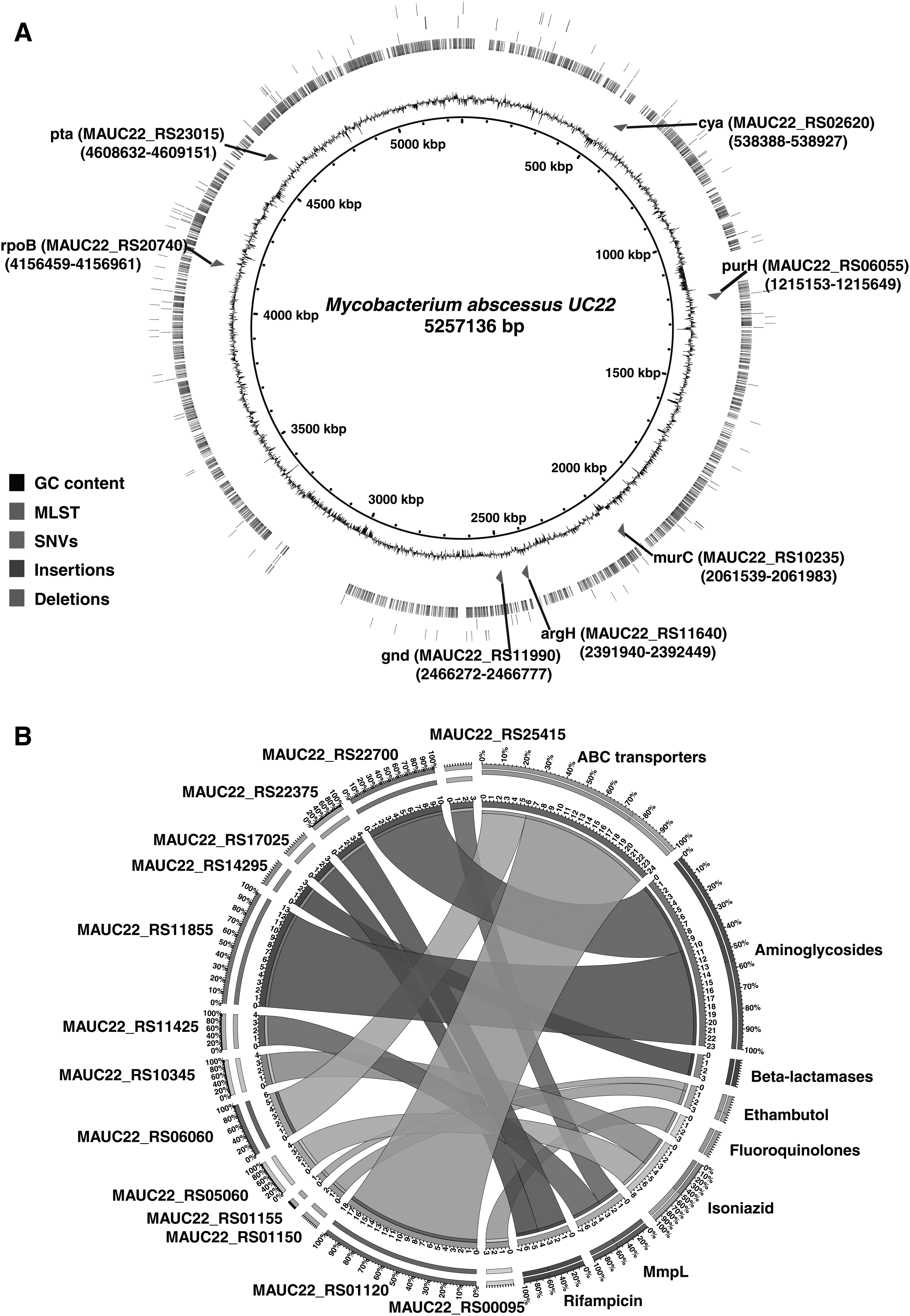
We carried out MLST on M. abscessus UC22 isolate and compared our MLST results with PubMLST database (M. abscessus complex MLST databases) to identify the allelic profile of the UC22 isolate (Jolley and Maiden, 2010). We found our isolate to share six identical allelic types of the seven (argH, cya, glpK, gnd, murC, pta, and purH) housekeeping genes used in MLST for M. abscessus. Our isolate was found to belong to sequence types (STs)-12 profile (Fig. 2A). The MLST approach is used to identify, classify, and assess the spread of epidemic clones throughout the world. The combination of alleles obtained at each locus defines its allelic profile or ST. The number of allelic types of housekeeping genes identified in the Mycobacterium abscessus UC22 isolate is shown in Table 1.
Studies have revealed M. abscessus to have an open pan-genome, which is rapidly evolving as opposed to M. tuberculosis. Horizontal gene transfer and homologous recombination phenomenon are known to be scarce in Mycobacterium sp. However, genome sequencing studies on M. abscessus have shown acquisition of virulence genes from other mycobacterial and nonmycobacterial species. Studies using MLST have also shown that some M. abscessus clinical isolates contain a genetic pattern displaying an amalgamation of alleles from the housekeeping genes corresponding to different subspecies (Macheras et al., 2014). This suggests that M. abscessus undergoes homologous recombination and horizontal gene transfer.
As there are not many studies published on M. abscessus clinical isolates from the Indian subcontinent, there is a lack of information on the prevalence and geographical distribution of the UC22 allelic profile. This warrants further need for multiomics-based studies on large numbers of clinical isolates of M. abscessus UC22 strain.
Identification of variants associated with natural and acquired resistance
To identify the genetic factors that may be associated with natural resistance, we compared SNVs identified in M. abscessus UC22 isolate with the genomes of publicly available M. abscessus isolates (Bryant et al., 2016). We observed 227 SNVs corresponding to 98 genes associated with natural resistance in M. abscessus UC22 isolate (Supplementary Table S2A). When we compared our data with publicly available data from M. abscessus and M. abscessus subspecies clinical isolates, we found 180/227 SNVs common among all the isolates. We found 19/227 SNVs common between our isolate and M. abscessus clinical isolates and 4/227 SNVs common between our isolate and M. abscessus subspecies clinical isolates.
We also uniquely identified 24 SNVs in genes known to confer drug resistance. It has been previously shown that M. abscessus monooxygenase orthologs may be potentially involved in resistance to rifampicin (Ripoll et al., 2009). We identified 26 SNVs corresponding to 13 genes that belong to the monooxygenase family in M. abscessus UC22 isolate. These SNVs have earlier been reported in several strains of M. abscessus. In addition, we also identified two SNVs (MAUC22_RS20735 and MAUC22_RS20740) in DNA-directed RNA polymerase subunit beta genes, which are known for conferring resistance to rifampicin. M. abscessus genome also encodes an Ambler class A β-lactamase gene, which shows homology with β-lactamases from the gram-negative bacteria (Nguyen and Thompson, 2006).
We observed three SNVs in class A β-lactamase gene (MAUC22_RS14295) (Supplementary Table S2A). The constant exposure of various mycobacterial strains to a large variety of β-lactams emanates mutations in their β-lactamase genes thus expanding their resistance spectrum even against the newly developed β-lactam antibiotic (Shaikh et al., 2015).
We observed 21 and 22 SNVs corresponding to 3 and 6 genes in aminoglycoside phosphotransferase proteins and N-acetyltransferase family of proteins, respectively (Supplementary Table S2A). In addition, we observed 114 SNVs in ABC transporter family and 17 SNVs in mmpL transporter family of genes (Fig. 2B; Supplementary Table S2A). M. abscessus also contains enzymes such as 2-N-acetyltransferase and aminoglycoside phosphotransferases that could alter aminoglycoside drugs by transferring acetyl or phosphate residues on key positions within the antibiotic and making them inactive (Nessar et al., 2012; Ripoll et al., 2009).
The ABC-type multidrug transporters act as importers or exporters by utilizing ATP energy to pump drugs out of the cell to the external environment (Kerr, 2002; Kerr et al., 2005). The mmpL transporter family belongs to resistance, nodulation, and cell division proteins, which are involved in lipid transport to the membrane with the help of proton motive force of the transmembrane electrochemical proton gradient (Goldberg et al., 1999).
We also compared SNVs associated with acquired resistance in our data with M. abscessus and M. abscessus subspecies isolates. In total, we identified 10 SNVs corresponding to 4 genes, of which SNV in 16S rRNA methyltransferase G gene (MAUC22_RS26020 A25P) was common in all the isolates (Supplementary Table S2B). We did not identify any SNVs in the 23S rRNA methyltransferase G gene (MAUC22_RS11405) associated with clarithromycin resistance, and thus, we expect the clinical isolate to be sensitive to clarithromycin (Mougari et al., 2017; Sohn et al., 2009).
Proteomic characterization of the M. abscessus UC22 strain
Following whole-genome sequencing analysis, we carried out LC-MS/MS-based proteomic profiling of M. abscessus UC22 isolate. Detailed workflow for proteomic analysis is illustrated in Figure 1. We identified 32,020 peptides corresponding to 3601 proteins, which is equivalent to ∼71% of the reference M. abscessus UC22 proteome. Of the total proteins, 686 and 35 proteins were uniquely identified in bRPLC and SCX fractions, respectively (Supplementary Fig. S1). The complete list of proteins and peptides identified in the current analysis is provided in Supplementary Table S3.
We further compared our data with the previously published proteomic study on M. abscessus ATCC 19977 strain (Miranda-CasoLuengo et al., 2016). Of the 2697 proteins reported by them, 1732 proteins were identified in the current analysis. Among these 1732, 1266 proteins have peptide coverage in both studies. However, we provide additional evidence for 466 proteins that was previously not reported, including TetR/AcrR family transcriptional regulator, WhiB family transcriptional regulator, and Tat subunit TatB (Supplementary Table S3).
TetR/AcrR family proteins are known to regulate a large number of activities, such as biosynthesis of antibiotics, multidrug resistance, efflux pumps, virulence, and pathogenicity of bacteria (Deng et al., 2013). TetR/AcrR family proteins mediate multidrug efflux pumps and might also serve as broad spectrum for new drug targets. M. abscessus genome consists of six whiB genes, which are known to be involved in conferring drug resistance to streptomycin, erythromycin, and tetracycline (Morris et al., 2005). The whiB family proteins are putative transcription factors involved in the regulation cellular processes such as pathogenesis, cell division, and responses to oxidative stress (Nessar et al., 2012).
Additional features of the UC22 strain proteome
Bacterial secretory proteins secreted through the general secretory (Sec) pathway are often found to have classical amino-terminal signal peptide sequences (Desvaux et al., 2010). SignalP analysis led to the identification of 293 proteins with signal peptides (Supplementary Table S3). These include hemophore-related proteins, iron transporters, and sensor domain-containing proteins. The subcellular localization analysis predicted 1991 (55%) proteins as cytoplasmic proteins, 790 (21%) localized to cytoplasmic membrane, 44 (1.2%) extracellular, and 14 (0.3%) localized to cell wall (Supplementary Table S3). Examples of proteins localized to the cytoplasm include TetR/AcrR family transcriptional regulator, acyl-CoA dehydrogenase, and α/β-hydrolase proteins.
Lipoproteins are varied class of secreted bacterial proteins that include 1–3% bacterial genome-encoding proteins (Babu et al., 2006). The PRED-LIPO tool predicted 121 lipoproteins in our proteomic data from M. abscessus UC22 clinical isolate (Bagos et al., 2008; Supplementary Table S3). These include sensor domain-containing proteins, Mce family of proteins, and ABC transporter substrate-binding proteins. Overexpression of ABC transporters in M. tuberculosis has been reported to contribute to resistance to several antibiotics (Wang et al., 2013). Similarly, ABC transporters are known for conferring natural resistance to several antibiotics in M. abscessus (Nessar et al., 2012).
Proteogenomic analysis
In the current study, using an integrated data analysis workflow, we carried out proteogenomic analysis M. abscessus UC22 strain.
Identification of novel genes
It is likely that computational pipelines miss a few bona fide protein-coding genes during genome annotation. Using the custom genome database, we identified seven potentially novel peptides of M. abscessus UC22, corresponding to seven novel protein coding genes (Supplementary Table S4A). We identified peptide evidence for a novel protein coding gene: IOB_NG_005, which encodes for a protein with 896 amino acids (Fig. 3A). An ortholog for the novel gene was identified with BLAST algorithm in M. abscessus subsp. abscessus strain 876, suggesting the conservation of this gene in M. abscessus.

Refinement of genome annotation of M. abscessus UC22 genome-based proteomic evidence.
We further performed SMART analysis to reveal mycobacterial membrane protein large (mmpL)—associated domain in this novel gene. The mmpL transporter family genes are distributed throughout the M. abscessus genome and these proteins mediate transport across the cytoplasmic membrane using the proton motive force of the transmembrane electrochemical proton gradient (Nessar et al., 2012). mmpLs are also known to play a role in the export of lipid components across the cell envelope, involved in physiology of mycobacteria and/or can act as virulence factors (Ripoll et al., 2009; Viljoen et al., 2017).
Correction of existing gene models
We identified six N-terminal extensions, which were also supported by ORFs, open reading frames predicted by FgeneSB and GeneMark (Supplementary Table S4B). N-terminal extension of MAUC22_RS08685 protein with one peptide mapping upstream of the gene is depicted in Figure 3B.
Confirmation of translational start sites
In this study, we carried out mass spectrometry-based proteomic approach to identify the modified N-terminal peptides. Using protein N-terminal acetylation as dynamic modification, 411 unique N-terminal peptides were obtained for 355 proteins reported in M. abscessus UC22 protein database. Among these proteins, 325 were confirmed with N-terminal peptides with the initiator methionine cleaved, and 30 were confirmed with initiator methionine residues (Supplementary Table S3). Correct assignment of translation start site is important for the protein sequence (Rison et al., 2007). Translational start sites (TSS) of a protein can be annotated by the identification of N-terminal acetylated peptides using mass spectrometry data (Zheng et al., 2017).
Earlier studies have reported that nontryptic nature at the N-terminus of the peptide, such as N-terminal peptides with an initiator methionine residue or an initiator methionine cleaved, could indicate the N-terminal of protein (Kelkar et al., 2011).
Utilization of proteogenomic approach to provide indispensible information for improvising genome annotation has previously been used to refine different mycobacterium genomes such as Mycobacterium smegmatis, M. tuberculosis H37Rv, and Mycobacterium vaccae. A previous study reported by Gallien et al. (2009), an ortho-proteogenomic approach was implemented, resulted in identification of 29 novel genes and confirmation of annotated start site for 342 proteins in M. smegmatis (Table 2). Another study by Potgieter et al. (2016) that focused on the proteogenomic analysis of M. smegmatis mc2155 strain identified 63 novel genes, 81 N-terminal extensions, and confirmation of annotated start site for 558 proteins.
A recent study on M. vaccae strain by Zheng et al. (2017) identified 38 novel genes, correction of 98 gene models, and confirmation of annotated start site for 445 proteins. Apart from these, a previous study published by our group revealed 41 novel genes, correction of 79 gene models, confirmation of annotated start site for 727 proteins, and correction of TSS for 33 proteins in M. tuberculosis H37Rv strain (Kelkar et al., 2011). Another recent study published by our group revealed 63 novel genes, correction of 25 gene models, and confirmation of annotated start site for 332 proteins in M. tuberculosis H37Ra strain (Pinto et al., 2018).
Identification of proteins encoded by the de novo assembled contigs
We identified 77 peptides corresponding to 24 contigs, which were not reported in M. abscessus UC22 reference proteome database. We identified five peptides that mapped to a protein encoded in the first forward strand of NODE_96 contig of our de novo assembled unmapped reads (Supplementary Table S5). Domain analysis with SMART showed that this protein contains p-aminobenzoate N-oxygenase (AurF) domain. We also confirmed the orthologus evidence of this protein in other strains of M. abscessus using the BLAST algorithm (Supplementary Fig. S2).
Similarly, four different peptides that mapped to protein encoded in the first forward strand of NODE_113 contig were also identified. Domain analysis of the identified protein revealed that it contains pyridoxamine 5′-phosphate oxidase domain, which is an FMN flavoprotein that catalyzes the oxidation of pyridoxamine-5-P (PMP) and pyridoxine-5-P (PNP) to pyridoxal-5-P (PLP). Earlier studies on M. tuberculosis have shown that pyridoxal 5′-phosphate biosynthesis is essential for survival and virulence (Ankisettypalli et al., 2016).
Using this approach, we thus provide the first direct protein-level evidence through identification of peptides mapping to contigs translated from the de novo assembled unmapped reads. These coding sequences have probably resulted from genomic rearrangements or gene duplications, which are not present in the M. abscessus UC22 genome or where coding regions vary in the genome M. abscessus UC22 clinical isolate.
Identification of variant peptides in M. abscessus UC22 proteome
A total of 553 variant peptides corresponding to 356 proteins were identified, of which 540 variant peptides had PSMs more than 2 (Supplementary Table S6). We further mapped the variants with SNVs in genes earlier reported to be associated with conferring drug resistance (Nessar et al., 2012). We identified 30 variant peptides corresponding to 16 genes carrying SNVs, resulting in rifampicin, isoniazid, aminoglycosides, and β-lactamase (Table 3). Our data, for the first time, provide evidence at the peptide level for SNVs conferring drug resistance in M. abscessus UC22 isolate (Fig. 4). It is interesting to note that of the 16 genes with variant peptides, 9 belonged to the ABC transporter gene family, which is involved in transporting multiple drugs across the membrane (Danilchanka et al., 2008).
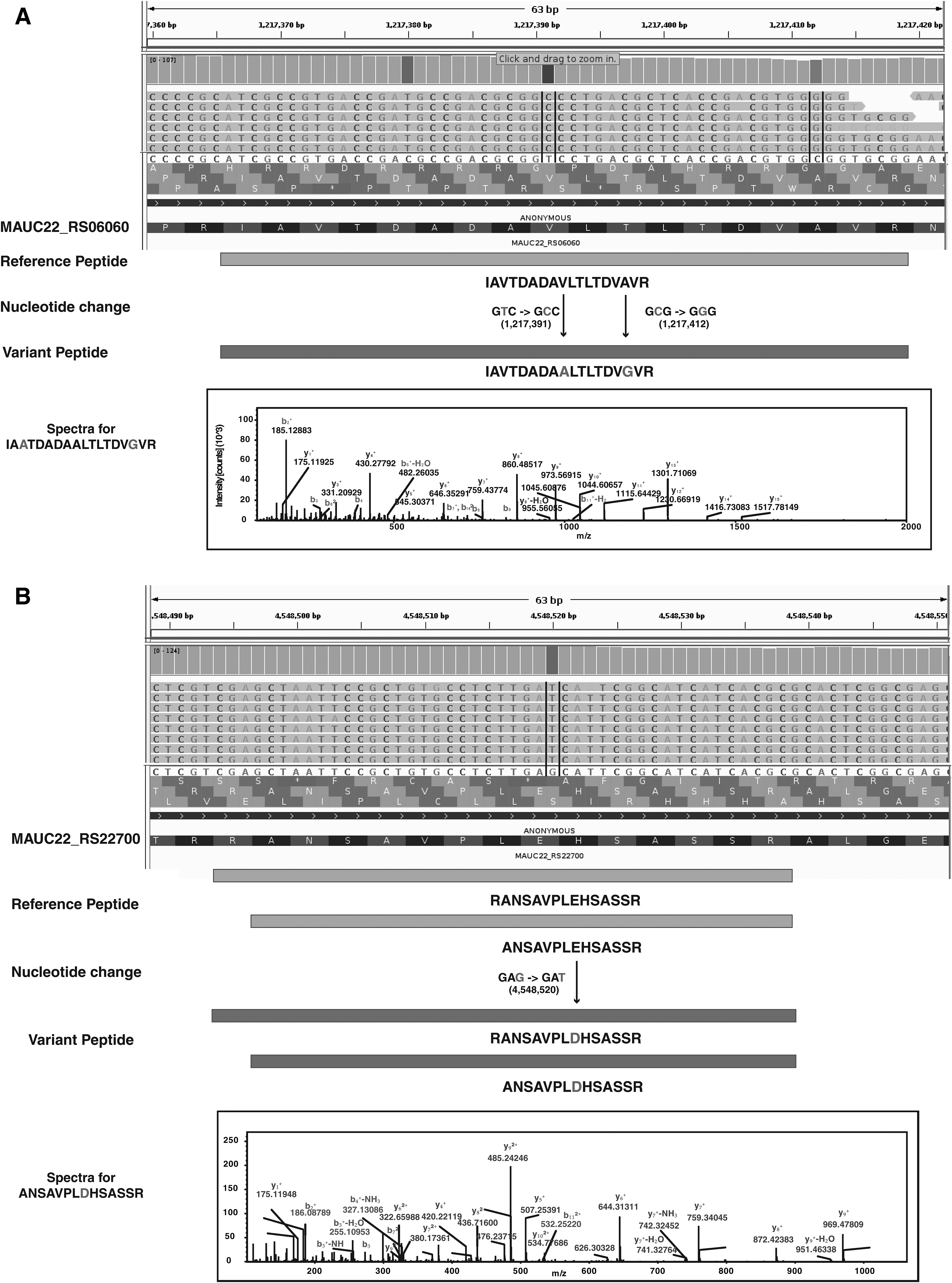
Representation of variant peptide evidence for SNVs identified in the M. abscessus UC22 clinical isolate.
PSM, peptide spectral match.
GO analysis of the mutated proteins in M. abscessus UC22 strain revealed them to be involved in catalytic activity (22%), metabolic process (25%), and DNA repair (4.6%) (Supplementary Table S6). These include methylated DNA—protein/cysteine methyltransferase, peptide synthetase, acetoacetyl-CoA synthetase, isopropylmalate isomerase, and 1-pyrroline-5-carboxylate dehydrogenase. Isopropylmalate isomerase enzyme is involved in leucine biosynthesis pathway.
An earlier study has shown that leucine biosynthesis pathway is necessary for the survival and virulence in M. tuberculosis (Manikandan et al., 2011). Previously published proteomic study on M. abscessus ATCC 19977 strain has shown that 1-pyrroline-5-carboxylate dehydrogenase (pruA) is involved in oxidation of proline to glutamate using carbon and nitrogen source for metabolism (Miranda-CasoLuengo et al., 2016). The GO-based functional annotation of identified proteins is depicted in Figure 5.
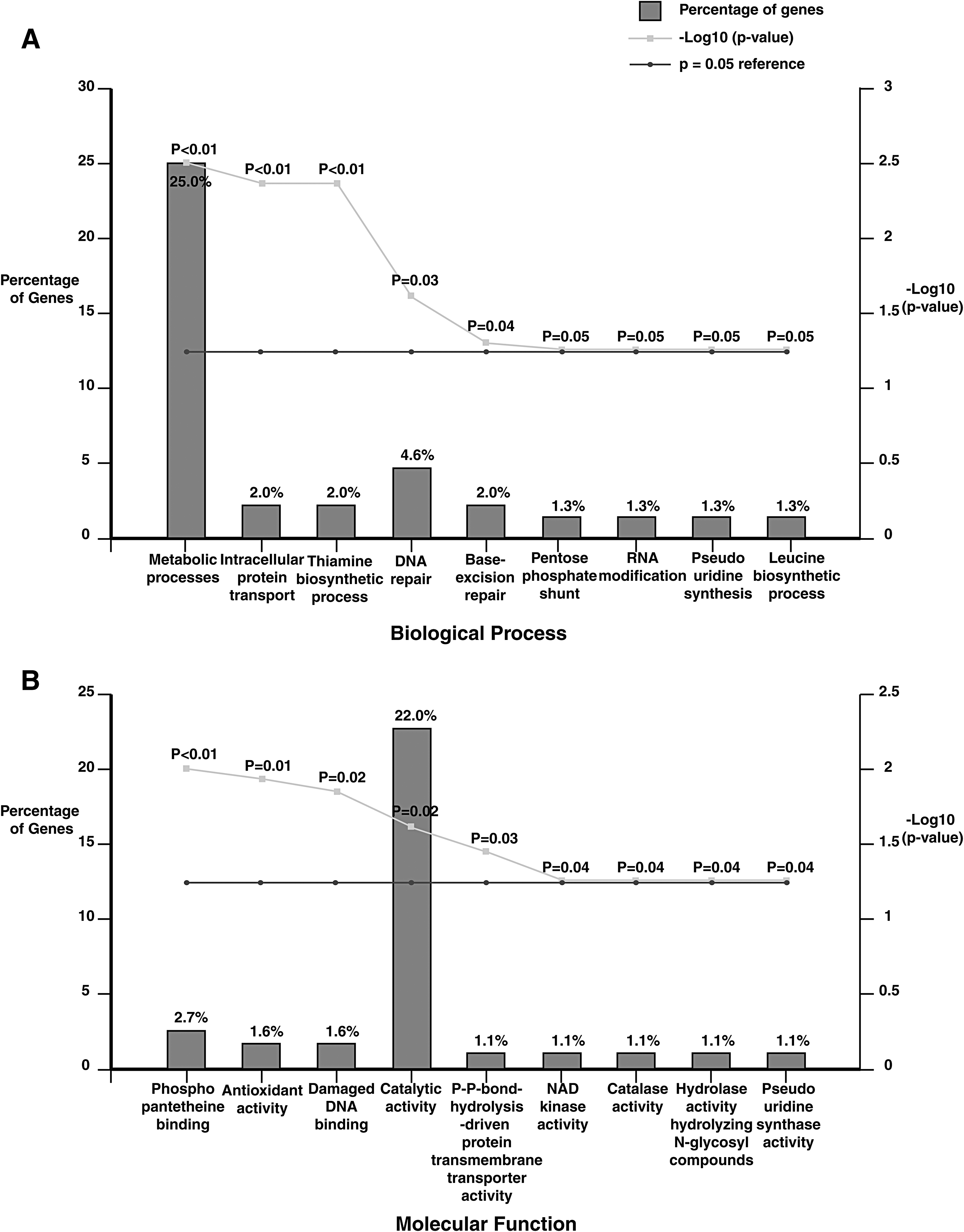
Gene ontology-based functional annotation of proteins carrying variant peptides.
Cell envelope proteins and virulence-associated genes
M. abscessus contains a large set of surface-exported Mce proteins organized in the genome (Ripoll et al., 2009). We found nine proteins belonging to Mce family in our proteomic data (Supplementary Table S7). We also found 14 SNVs corresponding to these 9 Mce proteins and variant peptide evidence for 3 of these SNVs in genome data (Supplementary Table S7). We have also identified these proteins in our lipoprotein prediction analysis. Mce family proteins of mycobacteria are known to be involved in the invasion of host macrophages and nonphagocytic mammalian cells (Zhang and Xie, 2011). In M. tuberculosis, Mce proteins have been shown to have a specific role in lipid transport (Cantrell et al., 2013; Forrellad et al., 2014).
Virulence factors play a very important role in promoting the growth of bacteria in the host environment and they also help in binding to the host cell for invasion (Choo et al., 2014). We identified 115 virulence genes in our proteomic data, which include ABC transporters, type VII secretion family of proteins, and RNA polymerase sigma factor protein family (Supplementary Table S7). These 115 virulence genes were distributed across different Mycobacterium species (Supplementary Fig. S3). Relapse of prolonged infections with the ability to persevere inside phagocytes is the major virulence mechanism known in M. abscessus (Pang et al., 2013). Our analysis reveals that virulence factors known to be involved in M. tuberculosis virulence have orthologs in the M. abscessus genomes of M. abscessus UC22 and M. abscessus ATCC 19977 strains (Table 4).
We identified integral membrane proteins (EccB, EccC, EccD, and EccE), which are present in most of the five ESX systems (Houben et al., 2012). We also identified EccA protein that belongs to the AAA + (ATPases associated with various cellular activities) protein family and is important for type VII secretion system (Houben et al., 2014). We have identified a similar set of integral transmembrane proteins in a comparative proteomic study of M. tuberculosis H37Ra and H37Rv strains published recently by our group (Verma et al., 2017). M. abscessus genome encodes for sigma factors known to be involved in M. tuberculosis virulence (Ripoll et al., 2009). We identified four of these sigma factors in our proteomic data set (SigE, SigF, SigL, and SigM).
We observed three SNVs in accessory Sec system translocase secA2 (MAUC22_RS11910), which are also a part of the virulence gene list with proteome and variant peptide evidence from the proteome data. SecA2 gene is essential to prevent phagosome maturation and also encodes a preprotein translocase ATPase that translocates superoxide dismutase (sodA). Studies have also shown that secA2 mutant of M. tuberculosis is attenuated for growth in macrophages and in a mouse model, which validates the importance of secA2 to M. tuberculosis virulence (Braunstein et al., 2003; Kurtz et al., 2006). We also identified sodA (MAUC22_RS21555) and nuoG (MAUC22_RS10600) in our proteome data, which are virulence factors in M. abscessus and are also found to be antiapoptotic factors (Choo et al., 2014).
Our analysis revealed many putative virulence factors from M. abscessus UC22 strain. At present, M. abscessus species is rapidly evolving and has already acquired antibiotic resistance. To understand the distribution of M. abscessus species, a large number of clinical isolates from the Indian subcontinent might have to be sequenced and examined. The candidate molecules identified in the current study could provide novel insights into biological mechanisms and pathogenesis of M. abscessus UC22 strain, when studied in the context of a large number of clinical isolates.
Conclusions
Clinical microbial proteogenomics provides an opportunity to identify universal candidates for therapeutic interventions by investigating the pace of changes incorporated in the genome and proteome of a pathogen across clinical isolates. This study integrates the genomic and proteomic data obtained from M. abscessus UC22 clinical isolate and provides insights on the strain-specific genomic and proteomic signatures. The proteogenomic approach along with our whole-genome sequencing analysis of UC22 strain helped map several novel genes and correct existing protein annotations.
Among other interesting findings are identification of putative virulence genes associated with the UC22 strain and peptide-level evidence for genomic variants. Our study shows the importance of integration of multiomics platforms for a deeper and better understanding of clinically significant pathogenic organisms.
Footnotes
Acknowledgments
The authors thank the Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka, for the support to the Center for Systems Biology and Molecular Medicine at Yenepoya (Deemed to be University) under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). The authors acknowledge Yenepoya (Deemed to be University) for access to mass spectrometry instrumentation facility. They thank the Department of Biotechnology (DBT), Government of India, for research support to the Institute of Bioinformatics.
J.A is a recipient of the Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India. R.V. is a recipient of the Senior Research Fellowship from University Grants Commission (UGC), Government of India. O.C. is a recipient of INSPIRE Fellowship from the Department of Science and Technology (DST). M.A.N. is a recipient of Junior Research Fellowship from University Grants Commission (UGC), Government of India. S.M.P. is a recipient of INSPIRE Faculty Award from DST, Government of India.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.