
Abstract
Abstract
MicroRNAs (miRNAs) serve as critical regulators of gene expression. However, their binding to target genes can be influenced by genetic variability within the miRNA-target interaction (MTI) sites. We performed an in silico sequence reanalysis to identify novel sequence variants within MTIs with potential functional impacts. A literature search of the PubMed and the Web of Science spanning the years 2008 to April 2018 identified 240 articles reporting MTIs in humans. Sequence reanalysis of reported MTI regions was performed using the Ensembl browser. We found 76 sequence variants within 23 MTIs. We present description of MTIs wherein sequence variants are present within both the mature miRNA seed region and the miRNA target, which we termed miR-gene-target-single nucleotide polymorphism (miR-GenTar-SNP). To the best of our knowledge, this is the first report on copresence of sequence variants within both miRNA gene and the target site. In the course our analyses, the need for extension of current terminology emerged and therefore, novel terminology was introduced: miR-indel, miR-double nucleotide polymorphism (DNP), miR-TS-indel, and miR-TS-DNP. Identification of novel MTI sequence variants is a hitherto understudied, but critical dimension in understanding the complexity of interactions and gene deregulation in various complex diseases. Because such variations might profoundly affect miRNA function, they should be taken into consideration in future research that depends on “variability science” such as precision medicine, human genetics, and genomics in the study of complex diseases. The findings presented herein offer a baseline for further systematic reanalysis of all reported MTIs in human and other species.
Introduction
M
It has been estimated that on average, one miRNA regulates expression of 100–200 target genes (Krek et al., 2005; Lim et al., 2005) and thus plays an important role in repression or activation of protein translation, reviewed in Kunej et al. (2012). miRNAs are involved in a variety of biological processes, such as cell growth, differentiation, apoptosis, and tumorigenesis (Iorio et al., 2010; Piletič and Kunej, 2016). Their function on downstream targets is achieved through the establishment of miRNA-target interaction (MTI). Pairing is based on the thermodynamics of binding and Watson-Crick complementarity between mature miRNA seed region, which can be found between nucleotides 2 and 8 from the 5′ end of the miRNA, and target site within mRNA (Chen et al., 2008; Salzman and Weidhaas, 2013).
However, binding of miRNA to its target gene can be interfered by a characteristic secondary structure of the target mRNA (Niu et al., 2015) and by several cis-deregulatory mechanisms, including chromosomal alterations, epigenetic modifications, and sequence variants (Sethupathy and Collins, 2008). Sequence variants within miRNA gene can affect its structure, transcription rate, stability, processing efficiency, and maturation, while sequence variants within target gene can alter the existing or create illegitimate target sites, and alter the content of a target gene (Georges et al., 2007).
Moreover, sequence variants that are found within MTIs, also referred to as polymorphisms in miRNAs and their target sites (PolymiRTS) (Bhattacharya et al., 2014), were often found to have a profound impact by interfering with miRNA function through destabilization of interaction and are thought to affect phenotypic variation, chemosensitivity in cancer, and disease susceptibility and development (Acunzo et al., 2015; Calin et al., 2004; Salzman and Weidhaas, 2013). Thus, understanding the role of sequence variants could have a tremendous potential in pharmacogenomics, molecular epidemiology, and individualized medicine (Mishra and Bertino, 2009).
Different classifications of miRNA sequence variants have been proposed previously (Georges et al., 2007; Mishra and Bertino, 2009; Obsteter et al., 2015). One of them suggests three major categories, including sequence variants affecting miRNA biogenesis, sequence variants in miRNA target sites, and sequence variants altering epigenetic regulation of miRNA genes (Mishra and Bertino, 2009).
MTI sequence variants have also been categorized according to the position within miRNA regulome. One of the proposals suggests classifications of sequence variants according to the location within miRNA genes (miR-single nucleotide polymorphisms [SNPs]), miRNA target sites (miR-TS-SNPs), or miRNA silencing machinery (SM) (Georges et al., 2007), which was later termed as miR-SM-SNPs (Obsteter et al., 2015). This classification was extended with a class of sequence variants residing within regulatory regions (miR-rSNPs), for example within transcription factor binding sites (Obsteter et al., 2015). Sequence variants within miRNA SM can be further distinguished whether they reside within genes encoding components of the SM or within miRNA genes overlapping Drosha and Dicer cleavage (Georges et al., 2007; Obsteter et al., 2015).
In addition to SNPs, sequence variants encompassing 2 bp or more, including insertions/deletions (indels) and consecutive SNPs, can be frequently found within MTI and are predicted to have a significant downstream effect on gene expression (Bhattacharya et al., 2014). One such example is the seed region of bta-miR-2489, in which the analysis of miRNA genetic variability in livestock species revealed double-nucleotide polymorphism (DNP) (Jevsinek Skok et al., 2013).
An important feature of sequence variants for its clinical significance and evolutionary studies is its minor allele frequency (MAF) value, which represents the second most frequent allele value in population. While many of the sequence variants occurring in the genome are neutral and, thus have no effect on phenotype, some are associated with positively selected long-range haplotypes and characteristic phenotypes (New SNP Attributes, NCBI, 2004; Saunders et al. 2007).
The number of research articles reporting MTIs has been increasing over the past few years. In addition, several novel sequence variants have been deposited in genomics databases recently. Several study approaches are implemented in articles reporting MTI, sequence variants, or both; however, the majority of articles identify sequence variants in gene regions, which are frequently aberrant in pathologies, and later conclude their effect on phenotype can be achieved through disrupted binding between miRNA and its target gene (Dzikiewicz-Krawczyk et al., 2014; Nicoloso et al., 2010; Niu et al., 2015; Richardson et al., 2013).
In some cases, connections have been made between deposited SNPs and experimentally validated MTIs using in silico sequence reanalysis in various phenotypes, for example, profiling of miR-TS-SNPs in leukemia (Doss et al., 2013), diabetes mellitus (Ghaedi et al., 2016), and analysis of sequence variants within miRNAs involved in aerobic glycolysis (Suresh et al., 2016). However, there is an urge for a systematic research to identify sequence variants in previously reported MTIs by overlapping genomic regions of MTIs with SNPs deposited in databases. Thus, previously reported data can be connected at novel levels, reducing time and costs of an experiment.
The aim of this study was to perform an in silico sequence reanalysis of genomic locations of previously reported MTIs to find sequence variants residing within previously reported interaction sites. Our analysis also revealed a lack of suitable terminology for description of MTI sequence variants. Therefore, we extended the existing terminology and systematically categorized sequence variants within MTIs.
Materials and Methods
We retrieved research articles from PubMed (http://ncbi.nlm.nih.gov/pubmed) and Web of Science (http://apps.webofknowledge.com) using the keywords, such as “microRNA, miRNA, miR, target, gene, target site, interaction, polymorphism, SNP, genetic variant, 3′UTR variant,” published between January 2008 and April 2018.
The inclusion criteria for the study were assessed based on four different criteria, including (i) the authors reported the exact genomic position of the interaction; (ii) the authors did not provide the position in the genome, but the listed nucleotide sequence of MTI could be easily found using TargetScan (http://targetscan.org) (Agarwal et al., 2015) to determine its genomic position; (iii) the authors did not provide the position in the genome, neither was the MTI listed in TargetScan, but the authors clearly delineated MTI and identified sequence variant within, which served as sufficient genomic information to map the MTI to its location in the genome; or (iv) the exact genomic information was not reported, but the MTI region was clearly delineated and could be found in Ensembl, based on reporting the nucleotide sequence of surrounding region.
In addition, only research articles reporting methods that are considered strong approaches for MTI validation (luciferase reporter assay, quantitative real-time polymerase chain reaction, and Western blot) (Chou et al., 2016) were selected for further analysis.
Overlap analysis of the reported MTIs and sequence variants was performed using Ensembl database, release 92 (http://ensembl.org) (Yates et al., 2016), which was also used to obtain MAF value. The terminology of reported miRNAs was standardized according to the latest miRNA nomenclature guidelines (Desvignes et al., 2015). The retrieved data are presented and complemented in accordance with the initiative for standardization of results presenting MTI (Piletič and Kunej 2017), which we adapted to fit to the presentation of miRNA genes. In addition, using PubMed, we ought to check whether the newly identified sequence variants within the reported MTIs have been previously shown to be associated with disease onset and progression.
Results
In this study, we performed in silico sequence reanalysis of previously reported genomic locations of experimentally validated MTIs, which revealed that several sequence variants are residing within the sites of interactions. Terminology regarding the miRNA-associated sequence variation classes is not yet complete; therefore, we have suggested some novel terms. With literature mining, we retrieved 240 articles reporting on MTIs in humans. These were further divided into review (40%) and research (60%) articles. Seventy (29% of retrieved articles) research articles published between January 2008 and April 2018 reported MTI with adequate genomic information for further in silico sequence reanalysis.
Overview of the retrieved MTI sequence variants using in silico sequence reanalysis
We analyzed 70 research articles reporting MTIs in humans and extracted data regarding the genomic location of MTI. Afterward, we performed in silico reanalysis of these genomic locations and found 23 examples of MTIs, where sequence variants can be found at the site of interaction. Our results are presented as a graphical summary in Figure 1 and more detailed information is presented in Supplementary Data.
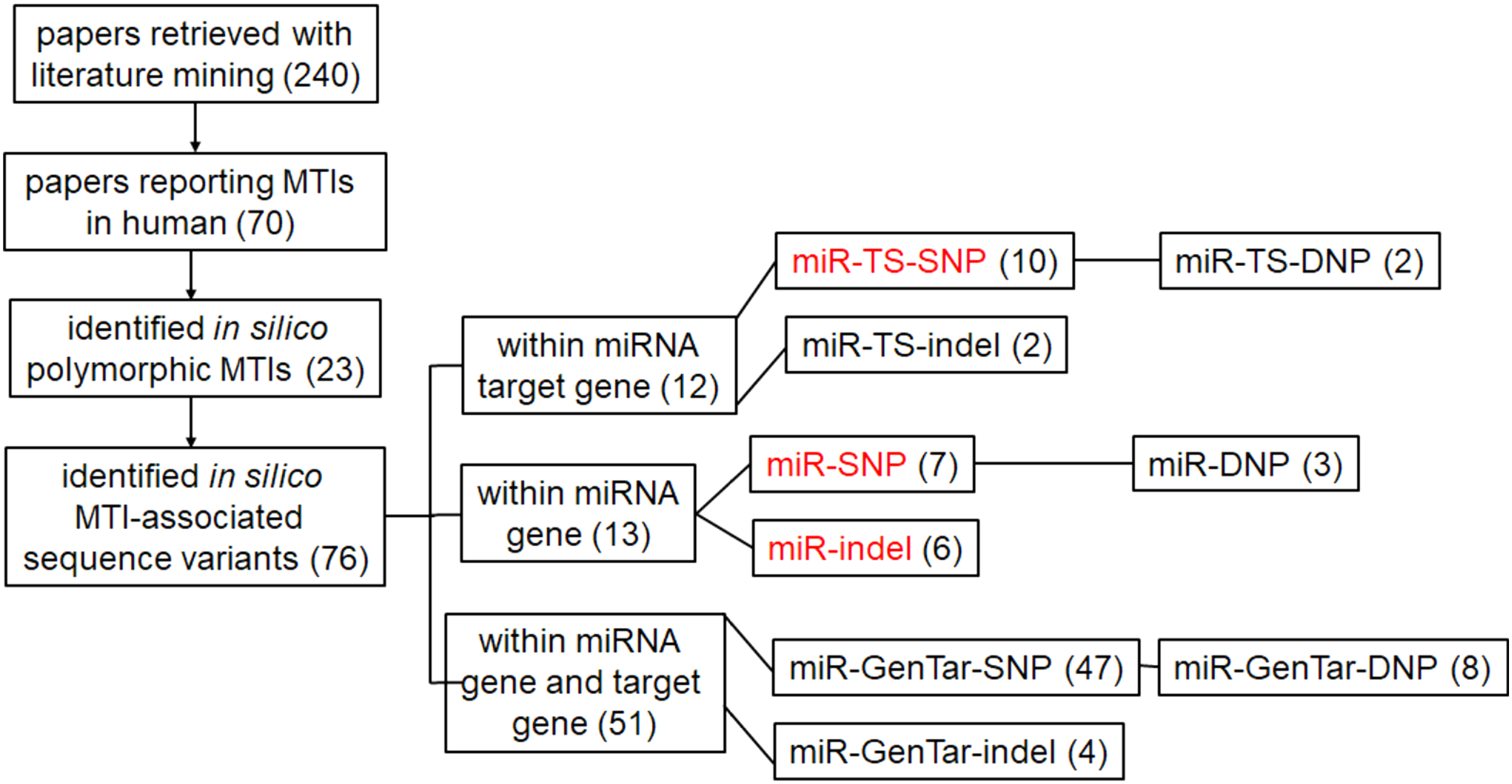
Graphical summary of results of in silico sequence reanalysis. The number of identified sequence variants is shown in brackets. Red color represents previously established acronyms, while black color represents suggested novel terminology for MTI sequence variants. MTI, microRNA-target interaction.
We present two main types of results. In the majority of cases (18), our result presents the first report of the presence of sequence variants within validated MTIs, while in some cases (5), it was possible to identify additional sequence variants within previously reported polymorphic MTIs, for example in the MTIs, reported in Dzikiewicz-Krawczyk et al. (2014), Liu et al. (2018b), Popp et al. (2016), Song et al. (2011), and Yang et al. (2017). Our analysis revealed 76 MTI-associated sequence variants residing within 23 previously reported MTIs, including 12 sequence variants within mature miRNA seed region, 13 sequence variants within the target gene, and 51 sequence variants within both: miRNA gene seed and target region.
Proposal for the extension of terminology describing MTI sequence variants
The identified novel types of MTI-associated sequence variants represent an urge to implement new terminology in the field of MTI research. Classification of miRNA sequence variants has already been suggested previously; however, based on the identified novel sequence variation classes of MTI sequence variants, we suggest the following three major categories to classify MTI sequence variants according to their position within miRNA regulome (Fig. 2):

Presentation of the analyzed MTI-associated sequence variants.
sequence variants within miRNA gene, including miRNA SNP (miR-SNP), miRNA DNP (miR-DNP), and miRNA insertion/deletion (miR-indel);
sequence variants within miRNA target sites, including miRNA target site SNP (miR-TS-SNP), miRNA target site DNP (miR-TS-DNP), and miRNA target site indel (miR-TS-indel);
sequence variants within both, mature miRNA seed region as well as within miRNA target site, including miR-gene-target-SNP (miR-GenTar-SNP), miR-gene-target-DNP (miR-GenTar-DNP), and miR-gene-target-indel (miR-GenTar-indel).
In accordance to the classification based on the position of sequence variants within miRNA regulome, the identified sequence variants fall into three subgroups. These include 13 sequence variants within mature miRNA seed region (miR-SNPs, miR-DNPs, and miR-indels), 12 sequence variants within target gene sites (miR-TS-SNPs and miR-TS-DNPs), and 13 examples of MTIs, where 51 sequence variants are present within both, mature miRNA seed region and miRNA target site (miR-GenTar-SNPs, miR-GenTar-DNPs, and miR-GenTar-indel).
According to the classification regarding sequence variant biotype, the identified sequence variants can be divided into three subgroups, including SNPs, DNPs, and insertions/deletions (indels). Among 76 MTI-associated sequence variants are 64 examples of MTI-SNPs (miR-SNPs, miR-TS-SNPs, and miR-GenTar-SNPs), out of which 13 examples of consecutive SNPs can be classified as MTI-DNPs (miR-DNP, miR-TS-DNP, and miR-GenTar-DNP), and 12 examples of indels (miR-TS-indel, miR-indel, and miR-GenTar-indel).
Identification of sequence variants within miRNA target genes
We identified several sequence variants residing within miRNA target genes in humans. Ten miR-TS-SNPs, including one miR-TS-DNPs, and two miR-TS-indel in humans were found within seven miRNA-target gene pairs. These sequence variants were identified within MTIs, previously validated in the following studies: MIR34C-5p/MIR449b-5p and ETV6, MIR589-3p and PML (Dzikiewicz-Krawczyk et al., 2014), MIR21 and CDC25A (Fu et al., 2015), MIR375 and ELAVL4 (Abdelmohsen et al., 2010), MIR155 and MAP3K10 (Wang et al., 2017), MIR1269A and TP53, and MIR1269A and CASP9 (Bao et al., 2018).
In one research article, the authors reported two polymorphic MTIs with one SNP within each of the MTI (Dzikiewicz-Krawczyk et al., 2014); however, using sequence reanalysis, we found an additional SNP within each of the previously reported polymorphic MTIs (examples MIR34C-5p/MIR449b-5p and ETV6, and MIR589-3p and PML). None of the identified sequence variants could be found, reported in the literature, to be involved in any particular disease, except for the sequence variants already reported to modulate risk of leukemias (Dzikiewicz-Krawczyk et al., 2014).
Identification of sequence variants within mature miRNA seed region
In addition to sequence variants found within miRNA target sites, our analysis revealed several sequence variants residing within mature miRNA seed region. We found seven miR-SNPs, including three miR-DNPs, were residing within three previously reported miRNA-target gene pairs: MIR302A and CCND1, MIR302C and CCND1, and MIR302D and CCND1 (Card et al., 2008). Within these three miRNA-target gene pairs, six cases of miR-indels were found. As with sequence variants within miRNA target genes, these sequence variants were also not reported to contribute to any disease.
Identification of MTI sequence variants copresent in miRNA and target gene
Reanalysis of previously validated MTIs also revealed 13 examples of MTIs, where SNPs and indels are present within both, mature miRNA seed region as well as miRNA target site (Fig. 3). To our knowledge, this is the first report of copresence of sequence variants within both miRNA gene and the target site. In the majority of cases, at least two sequence variants were identified within a target site, with single or multiple sequence variants residing on the pairing mature miRNA seed region.

Examples of sequence variants present within both, mature miRNA seed regions and miRNA target sites (miR-GenTar-SNPs, miR-GenTar-DNPs, and miR-GenTar-indels). Mature miRNA seed and miRNA target site sequences involved in MTIs are shown. Denoted in green color are sequence variants with their corresponding rs numbers and base change. DNP, double-nucleotide polymorphism; Indel, insertion–deletion; SNP, single-nucleotide polymorphism
In particular, we were able to identify 47 SNPs within those MTIs, including eight consecutive SNPs (DNPs), and four deletions. Importantly, out of these 51 sequence variants, only 5 miR-TS-SNPs have been already reported by authors (Dzikiewicz-Krawczyk et al., 2014; Liu et al., 2018b; Popp et al., 2016; Wang et al., 2018; Yang et al., 2017). Notably, some additional authors reported variants disturbing miRNA interaction with target through structural changes; however, these are residing outside validated MTI itself, and with our reanalysis, we could identify distinct sequence variants presented within MTI itself, which were not considered by the authors (Song et al., 2011; Zhang et al., 2018).
Interestingly, using in silico sequence reanalysis approach, we were able to identify up to eight sequence variants residing within a single MTI, namely MIR616 and PON1 (Wang et al., 2018). Other highly polymorphic MTIs where five or more (Saunders et al., 2007) sequence variants were found within both, mature miRNA seed region and miRNA target site, include MIR24-1 and SCN5A (Zhang et al., 2018), MIR2861 and MTHFR (Liu et al., 2018b), MIR449A and NFKBIA (Song et al., 2011), and MIR34C-5p and ETV6 (Dzikiewicz-Krawczyk et al., 2014).
We propose to name those types of sequence variants as miR-gene-target-SNP (miR-GenTar-SNP), miR-gene-target-DNP (miR-GenTar-DNP), and miR-gene-target-indel (miR-GenTar-indel), which should be used for describing MTIs, in which sequence variations are present within both, mature miRNA seed region and miRNA target site region. Strikingly, our sequence reanalysis revealed four examples of MTIs, where sequence variants in miRNA and target gene occur at exactly the same position, namely MIR342 and IGF1R (Liu et al., 2018a), MIR2861 and MTHFR (Liu et al., 2018b), MIR616 and PON1 (Wang et al., 2018), and MIR24-1 and SCN5A (Zhang et al., 2018).
These examples suggest that nucleotide base pairing might be strongly impaired and would possibly have an effect on phenotype; however, these assumptions have to be experimentally validated. Notably, analysis of newfound sequence variants within MIR24-1 and SCN5A binding pair (Zhang et al., 2018) revealed that all five identified sequence variants within miRNA target site, which were not reported by authors, associate with cardiac abnormalities, such as Brugada syndrome, sick sinus syndrome, and (congenital) long QT syndrome. These drastic changes are probably due to the location of MTI, where MIR24-1 binds to the last exon of SCN5A, causing frameshift and missense variants leading to truncated protein.
Allele frequency of identified sequence variants
MAF values are based on the frequency of the second most frequent allele in population and were retrieved to determine if the functional effect of sequence variant could compromise the interaction between miRNA and its target gene.
The majority of sequence variants included in this study are either observed in <1% in the population or lack population frequency information in Ensembl database. Ten out of eleven sequence variants in target sites have frequency information for various panels of human populations. Fifty percent of the observed population carry 2 out of 10 miR-TS-SNPs, while seven have MAF <0.01. Eleven out of thirteen sequence variants in miRNA genes have listed frequency information, with the most common variant present in 2% of the studied population. Forty out of fifty-one miR-GenTar-SNPs/miR-GenTar-indels have listed MAF value, out of which 34 have MAF value <0.01. One of the identified miR-GenTar-SNPs is present in 50% of the population, two in 17/18%, and three in 1%.
Discussion
A variety of different sequence variant classes were identified using in silico reanalysis of previously reported MTIs. Reanalyzing 70 research articles, we found sequence variants within 18 MTIs that were previously reported to be nonpolymorphic and identified additional sequence variants within five previously reported polymorphic MTIs. Seventy-six MTI-associated sequence variants were found, including 10 miR-TS-SNPs, out of which two can be classified as miR-TS-DNPs, 2 miR-TS-indel, seven miR-SNPs, including 3 miR-DNPs, and 6 examples of miR-indels. Interestingly, novel class of sequence variants emerged after reanalysis, as we found 47 miR-GenTar-SNPs, including eight examples of miR-GenTar-DNPs, and four examples of miR-GenTar-indel.
Identification of sequence variants within both mature miRNA seed region and miRNA target site emphasizes the importance of further data retrieval and recycling for systematic reanalysis of the field. Sequence variants were additionally checked for their MAF value and involvement in pathologies.
We performed a screening for sequence variants, which would reside in both, mature miRNA seed region and miRNA target site. Our analysis revealed 13 such examples, where we were able to identify up to eight sequence variants residing within a single MTI. As this is the first report on those types of sequence variants, we named them miR-GenTar-SNP, miR-GenTar-DNP, and miR-GenTar-indel. Since those sequence variants are residing within both interaction partners, we are assuming they have an important impact on miRNA binding with its target gene, thus further emphasizing the complexity of miRNA regulatory network.
To be able to further simplify this complexity, we also suggest novel classification of MTI sequence variants and propose novel terminology, which should be used to clarify the reporting on sequence variants residing within MTIs. The majority of research articles use term “polymorphisms” instead of using “sequence variant,” as suggested by HUGO Gene Nomenclature Committee (http://genenames.org) (Yates et al., 2017). In addition, so far the terminology has only distinguished between two types of sequence variants, miR-SNP and miR-TS-SNP, which are both based on “SNP” term. However, our study revealed that additional names and abbreviations for MTI sequence variants should be implemented, including miR-indel, miR-DNP, miR-TS-indel, and miR-TS-DNP.
Sequence variants within interaction site may either abolish or weaken miRNA target regulation, or create an illegitimate binding site, and are possibly a subject to selection. However, for a sequence variant to create a functional illegitimate binding site, the temporal and spatial coexpression of appropriate cognate miRNA and mRNA or ncRNA is necessary (Saunders et al., 2007). Thus, their effect and biological relevance need to be further tested with carefully conducted experiments.
Many of the retrieved sequence variants have no or very low assigned MAF values, which could be due to either the novelty or rarity of the variant, since most of the common variants have been sampled by projects such as 1000 Genomes Phase 3 (Auton et al., 2015), Exome Sequencing Project (https://esp.gs.washington.edu/drupal/), and Exome Aggregation Consortium (Lek et al., 2016), and deposited in Ensembl.
In the majority of cases listed in this article, the variants are rarely occurring in the observed population and are likely neutral or a subject of negative selection. In contrast, others are more common among the population and might, therefore, be a subject of positive selection due to changing the expression level of a target molecule. However, despite having low assigned MAF values, identified sequence variants might still potentially be functional, but have to be first experimentally validated. One such example is the study by Liu et al., (2018b), where rs915014 within MTHFR gene has been functionally validated to be associated with increased risk of atherosclerosis and was suggested as an outcome biomarker for atherosclerosis patients, despite reported MAF value <0.01 in Ensembl database.
Sequence variants residing within mature miRNA seed region and target site have been associated with various diseases and they represent an important source of molecular biomarkers.
Interestingly, Zhang et al. (2018) reported the involvement of functional sequence variant rs1805126 in SCN5A terminal exon outside MIR24-1 and SCN5A binding pair to be associated with heart failure; however, we identified five other sequence variants residing directly within MTI (rs794728924, CD077699, rs760837591, CM107403, and rs199473319), which were not reported by authors, but were already found to be involved in the same phenotype. They were all previously reported to associate with various distinct cardiac abnormalities, such as Brugada syndrome, sick sinus syndrome, and (congenital) long QT syndrome (Itoh et al., 2010; Stenson et al., 2017; Ware et al., 2012), or deposited in ClinVar (Nykamp et al., 2017), which have been connected to mutations in heart's primary voltage-gated sodium channel, NaV1.5, encoded by SCN5A, and its interacting genes, leading to heart failure (Zhang et al., 2018).
These newfound sequence variants might have a big impact on phenotype due to causing frameshift and missense mutations in terminal exon of SCN5A, and might thus also represent potential molecular mechanism by which MIR24-1 could suppress SCN5A expression, resulting in disease phenotype. Therefore, in silico sequence reanalysis approach for newfound sequence variants within previously reported MTIs associated with certain disease phenotypes could further help to explain the sequence variant molecular function and mechanisms by which it is contributing to the disease phenotype. Furthermore, since they might play a detrimental role in miRNA-target binding and thus affect downstream function of miRNAs, experimentally validated MTIs should be carefully reanalyzed for presence of potentially functional sequence variants.
We believe that reported in silico sequence reanalysis of genomic locations of previously reported MTIs serves as a good example of a method to identify potentially functional sequence variants within MTIs. Although this approach does not represent a canonical function validation with experiments, this method represents an important novel approach toward possible functional impact evaluation based on reanalysis of already reported nucleotide sequence data. Implementation of this approach can, however, be a quite challenging task, as the authors often do not report adequate genomic information. This problem has already been recognized by our previous studies (Piletič and Kunej 2017; Slemc and Kunej 2016).
To address the problem of correct data interpretation and insufficiency of the recent versions of genomic browsers, we had to implement several different criteria when selecting eligible articles. In addition, addressing functional impact of sequence variants can also be challenging since many of the variants deposited in Ensembl database still have no associated functions.
Regardless, MTI studies should provide a starting point for further research using existing databases and/or carefully planned experiments, consisting of sequencing analysis of targetome's sequence variants. The sequence variant mapping could additionally provide a tool for studying gain or loss of functional miRNA-target sites and following phenotype studies. In addition, obtaining sequence variants within experimentally validated MTIs could enable development of tools for sequence variant identification for medical applications.
Some efforts toward retrieving sequence variants residing within MTIs in cancer genes have already been made (Landi et al., 2008); however, the existing catalogues need to be updated and supplemented with data from experimental validation. We believe that in the near future, a genome-wide screening for all existing miR-GenTar-SNPs and miR-GenTar-indels should be conducted.
Conclusions
MTI sequence variants represent an important level of precise gene expression regulation and should, therefore, be taken into consideration, especially in the field of “variability science” such as precision medicine, human genetics, and genomics in the study of complex diseases. Our reanalysis revealed several sequence variants within previously reported MTIs that were not reported by the authors, which could possibly affect the outcome of research. In addition, using in silico sequence reanalysis, we were able to identify potentially functional sequence variations that might contribute to molecular mechanism of pathologies. To our knowledge, this is the first report of copresence of sequence variants within both miRNA gene and the target site.
In the future, a systematic study of all reported experimentally validated MTIs should be conducted to identify MTI sequence variants. In addition, miR-GenTar-SNPs and miR-GenTar-indels need to be further analyzed to determine their effect on (disease) phenotype. The novel approach toward possible functional impact evaluation could now also be used for molecular studies of MTI–miRnome and in silico resequencing of other types of molecular interactions.
Identification of novel MTI sequence variants is a hitherto understudied, but critical dimension in understanding the complexity of interactions and gene deregulation in various complex diseases. Because such variations might profoundly affect miRNA function, they should be taken into consideration in future research that depends on “variability science” such as precision medicine, human genetics, and genomics in the study of complex diseases. The findings presented herein offer a baseline for further systematic reanalysis of all reported MTIs in humans and other species.
Footnotes
Acknowledgment
This study was funded by the Slovenian Research Agency (ARRS) through the Research Program Comparative genomics and genome biodiversity (grant number P4-0220).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.