
Abstract
Abstract
Eye disorders and resulting visual loss are major public health problems affecting millions of people worldwide. In this context, the sclera is an opaque, thick outer coat covering more than 80% of the eye, and essential in maintaining the shape of the eye and protecting the intraocular contents against infection and the external environment. Despite efforts undertaken to decipher the scleral proteome, the functional and structural picture of the sclera still remain elusive. Recently, proteomics has arisen as a powerful tool that enables identification of proteins playing a critical role in health and disease. Therefore, we carried out an in-depth proteomic analysis of the human scleral tissue using a high-resolution Orbitrap Fusion Tribrid mass spectrometer. We identified 4493 proteins using SequestHT and Mascot as search algorithms in Proteome Discoverer 2.1. Importantly, the proteins, including radixin, synaptopodin, paladin, netrin 1, and kelch-like family member 41, were identified for the first time in human sclera. Gene ontology analysis unveiled that the majority of proteins were localized to the cytoplasm and involved in cell communication and metabolism. In sum, this study offers the largest catalog of proteins identified in sclera with the aim of facilitating their contribution to diagnostics and therapeutics innovation in visual health and autoimmune disorders. This study also provides a valuable baseline for future investigations so as to map the dynamic changes that occur in sclera in various pathological conditions.
Introduction
The sclera is an opaque, thick outer coat covering more than 80% of the eye, and essential in maintaining the shape of the eye and protecting the intraocular contents against infection and the external environment. Despite efforts undertaken to decipher the scleral proteome, the functional and structural picture of the sclera still remain elusive.
The human sclera comprises episclera, scleral stroma, and lamina fascia (Coudrillier et al., 2015; Yoshida et al., 2014). It primarily consists of type I and III types of collagen, along with a lower amount of collagens IV, V, VI, VIII, XII, and XIII (Shelton and Rada, 2009). The extracellular matrix also consists of elastin, proteoglycans, and noncollagenous glycoproteins (Frost and Norton, 2007; Liu et al., 2017; Marshall, 1995; Rada et al., 2006). The extracellular matrix of sclera is synthesized by the fibroblasts that are located in between the irregularly arranged lamellae (Rada et al., 2006). The scleral thickness is not uniform throughout and its rigid structure prevents the fluctuation of intraocular pressure, facilitating rotation of the eyeball, while maintaining the optical stability.
In the postgenomic era, proteomics has emerged as an indispensable tool in providing valuable insights into the pathogenesis of common eye-related disorders, which could eventually result in the development of new therapies and treatment modalities. In-depth proteomic profiling of scleral tissue can provide valuable insights into its normal physiology.
Several investigations have been carried out to define the molecular profile of different regions of the eye in normal physiology and disease. The availability of high-resolution mass spectrometry has now enabled the deeper mining of the human proteome (Kim et al., 2014).
The Human Eye Proteome Project (HPP) was founded by the Human Proteome Organization (HUPO) in September 2012 with a goal of defining the eye proteome (Semba et al., 2013). Our group has been actively involved in elucidating the proteomes of various ocular tissues such as retina (Kim et al., 2014); ciliary body (Goel et al., 2013); vitreous humor (Murthy et al., 2014); aqueous humor (Murthy et al., 2015); iris (Murthy et al., 2016); and choroid (Dammalli et al., 2017). Various techniques have been used in the past by different groups to characterize the proteome of sclera as highlighted in Table 1. However, comprehensive proteomics studies are still required for an in-depth understanding of visual health in health and disease.
A Summary of Previously Published Studies for the Human Sclera Tissue Protein Identification
In this study, we carried out a proteomic analysis of sclera using high-resolution tandem mass spectrometry (MS/MS). We have also determined the relative abundance of proteins in the sclera. The data obtained from this study serve as a valuable platform to compare the changes in protein expression patterns occurring in other sight-threatening pathological diseases of the sclera.
Materials and Methods
Subjects, sample collection, and storage
Three scleral tissues were procured from healthy donor eyes after obtaining approval from the institutional ethics review board of the Vittala International Institute of Ophthalmology, Bangalore, India, and in accordance with the tenets of the Declaration of Helsinki. The right eye was selected from one donor adult (female) and left eye from two donor adults (two males), with no history of eye disease or previous eye surgery. The specimens were frozen and stored at −80°C until further analysis. The details of the tissues used are provided in Supplementary Table S1.
Preparation of samples for protein identification
The sclera tissues were homogenized using liquid nitrogen and lysed in lysis buffer containing 4% sodium dodecyl sulfate (SDS) in 50 mM triethylammonium bicarbonate (TEABC). The samples were then subjected to sonication thrice for 10 sec. Sonication of samples was carried out after placing samples on ice to prevent overheating during the process. The sonicated samples were then heated at 90°C for 5 min. The lysates were cooled at room temperature and centrifuged at 12,000 rpm for 10 min. The concentration of protein present in the supernatant was determined using the bicinchoninic acid assay kit (Thermo Scientific Pierce), where 150 μg of protein was subjected to in-gel digestion, while 300 μg of protein aliquot was used for in-solution digestion.
In-gel digestion
Approximately, 150 μg of protein was resolved on a 10% SDS-PAGE gel. The gel was stained with colloidal Coomassie blue, and 24 bands were cut from the lane containing the sample. In-gel digestion was carried out for these protein gel bands as described earlier (Balakrishnan et al., 2014). The reduction was carried out using 5 mM dithiothreitol (DTT) at 60°C for 1 h to reduce the disulfide bonds of the proteins. The samples were treated with 20 mM iodoacetamide (IAA) for 10 min at room temperature in the dark to alkylate the reduced bonds. Sequencing grade-modified trypsin (cat. no. V5111; Promega, Madison, WI, USA) was dissolved in TEABC and added to the gel pieces at an enzyme:substrate ratio of 1:20 and digestion was carried out at 4°C for 1 h.
Excess amounts of trypsin were removed, and the gel pieces were incubated at 37°C in 20 mM TEABC overnight. Peptides were extracted using 80% can and 0.5% acetic acid and dried. They were then reconstituted in 0.1% formic acid and desalted using C18 StageTips (3 M Empore high-performance extraction disks) before mass spectrometric analysis (Pinto et al., 2015). The dried peptides were stored at −80°C until liquid chromatography-MS/MS (LC-MS/MS) analysis.
In-solution trypsin digestion
Briefly, 300 μg protein from the pooled samples was reduced by incubating in 10 mM DTT at 60°C for 20 min. Alkylation was carried out in the dark with 20 mM IAA at room temperature for 10 min. The lysate was further subjected to acetone precipitation, and the pellet was dissolved in 50 mM TEABC. Digestion was carried out at 37°C for 16 h using L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)-treated trypsin (Worthington Biochemical Corporation, Lakewood, NJ, USA) at a final concentration of 1:20 (w/w). The reaction was acidified using 0.1% formic acid, and the peptides were lyophilized and stored at −80°C until further use.
Basic RPLC-based fractionation
Basic reverse phase liquid chromatography solvent A (10 mM TEABC buffer, pH ∼8.5) was added to the in-solution digested peptides and loaded onto a Waters XBridge column (Waters Corporation, Milford, MA, USA; 130 Å, 5 μm, 250 × 4.6 mm) using a Hitachi LaChrom Elite HPLC system, maintaining a flow rate of 0.5 mL/min. The peptide separation was achieved using a 130-min gradient, at a flow rate of 0.5 mL/min of solvent A (10 mM TEABC buffer, pH ∼8.5) and B (10 mM TEABC buffer, 90% acetonitrile, pH ∼8.5).
The fractionation was continued at 97% solvent A for 20 min, followed by 3% solvent B for 0–5 min, 10% solvent B for 5–10 min, 10–35% solvent B for 10–40 min, and 100% solvent B for 40–45 min gradient. Flow-through fractions were collected using a 96-well plate from the 10th minute and was finally concatenated into 24 fractions. Pooled samples were lyophilized and stored at −80°C until they were subjected to LC-MS/MS analysis.
MS/MS analyses
LC-MS/MS analysis of the samples was analyzed using Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) interfaced with Easy-nLC-1200 (Thermo Scientific, Bremen, Germany). The peptides obtained after C18 cleaning were resuspended in 0.1% formic acid (Solvent A) and loaded onto the trap column (Thermo Scientific, 75 μm × 2 cm, nanoViper, 3 μm, 100 Å) filled with C18 at a flow rate of 4 μL/min. The peptides were further resolved onto an analytical column (Thermo Scientific EASY-Spray RSLC C18 2 μm 15 × 50 μm). Data were acquired in data-dependent acquisition mode at a scan range of 400–1600, and in positive mode with a maximum injection time of 55 msec using an Orbitrap mass analyzer at a mass resolution of 60,000.
Top 10 intense precursor ions were selected for each duty cycle and subjected to higher energy collision-induced dissociation with 33% normalized collision energy. The fragmented ions were detected using Orbitrap mass analyzer at a resolution of 120,000 with a maximum injection time of 200 msec. Lock mass was set as an internal calibration using polydimethylcyclosiloxane (m/z, 445.1200025) ions (Olsen et al., 2005).
Data processing of LC-MS/MS analyses
Mass spectrometry-derived data were searched against Human RefSeq 75 protein database (consisting of 36,713 entries along with common contaminants) in Proteome Discoverer 2.1 (Thermo Scientific) using Sequest HT and Mascot (version 2.5.1; Matrix Science, London, United Kingdom) search algorithms. The parameters included trypsin as a proteolytic enzyme with maximum two missed cleavages. Cysteine carbamidomethylation was specified as fixed modification and acetylation of protein N-terminus and oxidation of methionine were set as variable modifications with a minimum peptide length of seven amino acids. The precursor mass tolerance was fixed at 10 ppm, and 0.05 Da was set for fragment ion tolerance. The data were searched against the decoy database with a 1% false discovery rate cutoff at the peptide level.
Bioinformatics analyses
Bioinformatics analysis was carried out to assign functional roles to the proteins identified in the sclera. The proteins identified in our study were classified based on the biological process, subcellular localization, and molecular function using ProteinCenter Annotation node in Proteome Discoverer 2.1. Pathway analysis was performed using DAVID (The Database for Annotation, Visualization, and Integrated Discovery) bioinformatics resources 6.8 (https://david.ncifcrf.gov) (Huang da et al., 2009). Label-free quantitation was carried out where the relative abundance of the proteins was calculated using normalized spectral abundance factor (NSAF) and distributed NSAF (dNSAF) (Liu et al., 2004; Zhang et al., 2010; Zybailov et al., 2006). It provides an improved measure for relative abundance by taking into account the length of the protein where the protein length is used to normalize spectral count (Sc) for improving the accuracy.
where Sc = spectral count of protein J, L = length of the protein, and N = total number of proteins. The top 200 abundant proteins expressed in the sclera were used to generate a protein–protein interaction (PPI) network using Search Tool for the Retrieval of Interacting Genes (STRING) (www.string-db.org). K-means clustering was employed to cluster the network into 10 clusters, and disconnected nodes were set to hidden. Highest confidence interaction scores (0.900) were used to filter the interaction network. The generated network was then exported into Cytoscape 3.6.1 (Shannon et al., 2003) and network topology parameters were computed using Network Analyzer, using the undirected option. The betweenness centrality of nodes was used to plot the interaction network.
Data availability
The mass spectrometry data and the MSF files obtained from this study have been uploaded to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository (Vizcaino et al., 2016) with the dataset identifier PXD008999.
Results and Discussion
Proteomics analysis of human scleral samples was performed in an unbiased approach using multiple fractionation techniques, including in-gel and in-solution digestion, as illustrated in Figure 1. Mass spectrometry analysis of both in-gel and in-solution fractions was carried out on a high-resolution Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific). The corresponding MS data were searched against Human RefSeq 75 protein database consisting of 36,713 entries along with common contaminants using SequestHT and Mascot, which resulted in the acquisition of 695,218 MS/MS spectra, of which 225,758 peptide-spectrum matches were assigned to 21,164 peptides corresponding to 4493 proteins.
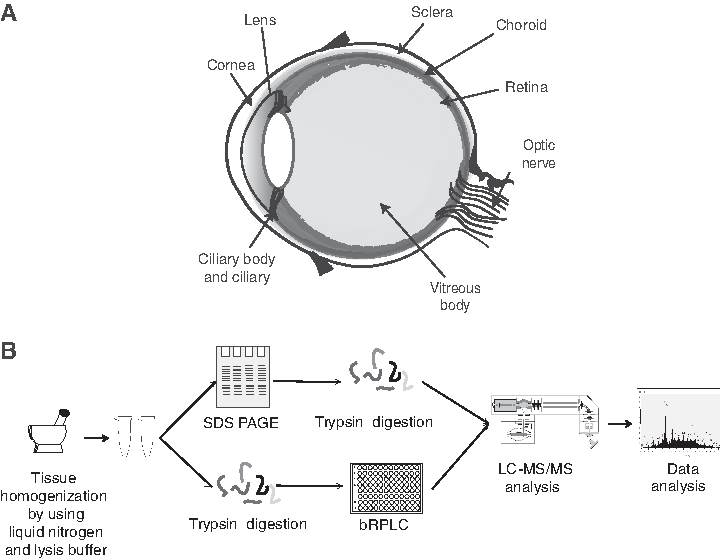
Schematic representation of the anatomical structure and proteomic analysis of human sclera tissue.
A list of proteins identified in the human scleral tissue is provided in Supplementary Table S2, of which 2931 proteins were found to be unique to this study after comparison with previously reported high-throughput studies on the human sclera (Table 1 and Supplementary Fig. S1). A list of all the identified peptides along with their corresponding proteins is provided in Supplementary Table S3.
Functional classification of proteins identified in the sclera
To understand the structural and functional importance of the scleral proteins, we carried out gene ontology (GO)-based classification of proteins identified from sclera using ProteinCenter Annotation node in Proteome Discoverer 2.1. Of the 4493 proteins identified, 4124 proteins were mapped to specific biological processes, 4214 proteins were assigned molecular functions, and 4225 proteins were related to diverse cellular components.
As illustrated in Figure 2A, the biological processes enriched included regulation of biological process (20%), followed by the metabolic process (20%), response to stimulus (15%), cell organization, and biogenesis (13%). Classification based on molecular function (Fig. 2B) revealed that a maximal number of proteins are involved in protein binding activity (34%). Classification of proteins by their subcellular localization revealed that the majority of proteins were localized to the membrane (21%), followed by cytoplasm (18%), nucleus (13%), and cytosol (13%), as depicted in Figure 2C.

Subcellular localization and functional annotation of proteins identified in sclera tissue, comparison with previous study and the pathway analysis.
Reported proteins in the human sclera
Among the proteins identified in our study, 1158 of them have been previously reported in a study by Zhang et al. (2016), authenticating the validity of our proteomics analysis. Some of the previously reported proteins in sclera include collagen type XII alpha 1 chain (COL12A1), thrombospondin 4 (THBS4), proline- and arginine-rich end leucine-rich repeat protein (PRELP), and biglycan (BGN).
COL12A1 belongs to the member of fibril-associated collagens with interrupted triple helices collagen family and is known to enhance the stability of the connective tissues (Bader et al., 2009). THBS4 is known to mediate cell to matrix interactions where its pentameric form allows binding of heparin and calcium. It is also known to be abundant in the trabecular meshwork and uveoscleral pathways and plays a role in the regulation of aqueous outflow resistance (Weissman et al., 2003). Similarly, PRELP is a leucine-rich repeat protein and known to be present in abundance in the human sclera (Johnson et al., 2006). Its high expression indicates its critical role in the biochemical regulation of scleral extracellular matrix and its active role in early development of the ocular growth in case of children and adolescents (Johnson et al., 2006).
Decorin (DCN) belongs to the member of small leucine-rich repeat protein (SLRP) family and plays a role in wound healing (Frikeche et al., 2016). As SLRP proteoglycans play a vital role in collagen fibrillogenesis, DCN is crucial for growth and repair of the sclera by increasing the fibril diameter (Neame et al., 2000). Phosphoglycerate kinase 1 is also reported in ocular surface epithelial regions, including the cornea, limbus, limbal epithelial crypt, and conjunctiva (Kulkarni et al., 2011). The representative MS/MS spectra for two of the previously studied proteins are shown in Figure 3A and B.

Representative MS/MS spectra of some proteins previously described in other studies.
Abundant proteins in the scleral proteome
NSAF method for each protein was calculated to estimate the abundances of proteins in the scleral proteome (Liu et al., 2004). Four proteins had NSAF value greater than 0.01, including albumin (ALB), prolargin (PRELP), vimentin (VIM), and histone cluster 1 H4 family member h (HIST1H4H) followed by actins, tubulins, collagens (COL12A1, COL6A3, COL6A3, COL6A1, COL6A2, COL6A2, COL1A1, and COL1A2), histone cluster proteins (HIST1H4H, HIST2H2BF, HIST1H2AD, HIST1H2AE, HIST3H2BB, HIST2H3D, HIST1H1C, HIST2H2AB, and HIST1H1E), and S100 calcium-binding proteins (S100A6, S100A4, S100A10, S100A11, S100A8, and S100A16) (Liu et al., 2004). The list of the proteins with their calculated NSAF and dNSAF values is provided in Supplementary Table S4.
We further carried out network analysis (Fig. 4 and Supplementary Tables S5–S7) of the top 200 most abundant proteins identified in sclera forming many clusters. PPI topology analysis showed the nodes with highest betweenness centrality comprising LUM (Lumican) and RhoA (ras homolog family member A). LUM has been known to regulate collagen fibrillogenesis and has been associated with axial myopia in humans (Yeh et al., 2010). This suggests its possible role during tissue repair and maintaining structural integrity. Mutations in LUM and DCN have also been associated with connective tissue disorders such as Stickler syndrome and Marfan syndrome resulting in visual difficulties (Young et al., 1998).
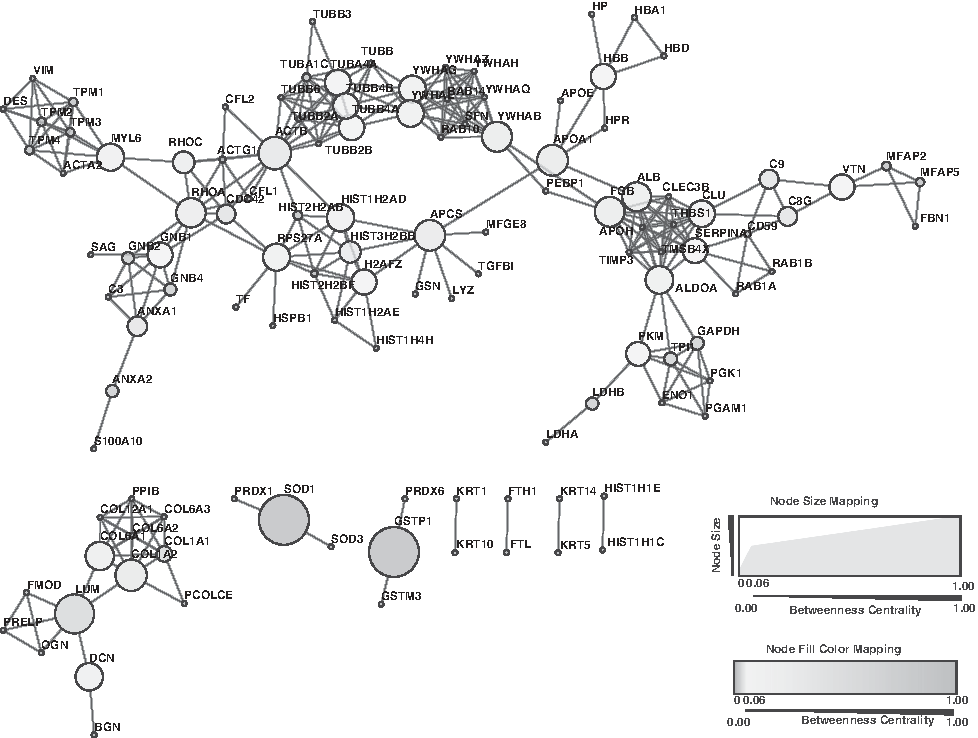
Interactome of the top 200 abundant proteins expressed in the human sclera. Network analysis of the abundant proteins was carried out using STRING (www.string-db.org). The network was then input into Cytoscape and network topology parameters were computed using NetworkAnalyzer. The betweenness centrality measure was used to depict the relationship between nodes.
In addition, we also identified RhoA, which plays an active role in the regulation of the cytoskeleton (Amano et al., 2010). There are reports suggesting its critical role in sclera remodeling and its activation induces sclera myofibroblast differentiation (Yuan et al., 2018). A previous study reported that the inhibition of trabecular meshwork RhoA pathway results in decrease of elevated nocturnal intraocular pressure (Borras et al., 2015).
SOD1 (superoxide dismutase 1) and GSTP1 (glutathione S-transferase P) were found to have high betweenness centrality, but were only connected to two nodes each. The relatively higher betweenness centrality scores assigned to these genes were due to the relatively smaller interaction network formed by these genes. SOD1 and GSTP1 have been reported to play an important role in protecting the ocular tissue from the toxicants produced during lipid peroxidation, hence protecting from degenerative ocular diseases (Behndig et al., 1998; Srivastava et al., 1994).
We also identified clusterin (CLU), which has been demonstrated to play a crucial role in biological events such as cell death, lipid transport, membrane integrity, and neurodegenerative disorders. It is reported to be expressed across different eye tissues such as ciliary body, cornea, retina, and aqueous and vitreous fluid, suggesting its importance in the pathophysiology of the eye (Wong et al., 2000). Its reduced expression results in the abnormal deposition of extracellular matrix, which is observed in the patients with pseudoexfoliation syndrome (Zenkel et al., 2006). We also identified an interesting cluster of 14-3-3 family of proteins (YWHAB, YWHAH, YWHAG, YWHAQ, YWHAZ, and SFN), which are known to be ubiquitous to the retina (Inamdar et al., 2018). Its expression in rod photoreceptors makes it vulnerable to neuropathy-associated stresses such as glaucoma (Ivanov et al., 2006).
Pathway enrichment analyses using DAVID
We carried out pathway analysis using the DAVID to map the proteins identified in the sclera (Fig. 2E and Supplementary Table S8). The most significantly enriched pathways included complement and coagulation cascades, focal adhesion, ECM-receptor interaction, glycolysis, and gluconeogenesis. The proteins identified in this study are highlighted by shading in Figure 5. We identified 41 scleral proteins to be involved in regulation of the actin cytoskeleton pathway. This pathway is a vital phenomenon, facilitating the formation of structures such as stress fibers and focal adhesions, and any malfunctioning of these proteins results in infections. Another significant pathway enriched in the sclera was the glycolysis pathway with 29 scleral proteins mapping to it, including alcohol dehydrogenases (ADH1B, ADH1C, ADH5, and AKR1A1), enolases (ENO1, ENO2, and ENO3), and hexokinase-1 (HK1).

Pathway representation of identified proteins involved in the regulation of the actin cytoskeleton. We identified 41 proteins to be involved in this pathway. These proteins are significantly enriched and may contribute to the contractile properties.
We also identified the distribution of the proteoglycans in the sclera such as DCN, BGN, LUM, prolargin, and mimecan. The extracellular matrix of sclera is predominantly composed of collagen, which undergoes remodeling during myopia causing significant thinning of the tissue. The integrin receptors identified in our study include alpha-1, alpha-2, alpha-3, alpha-4, alpha-5, alpha-6, alpha-7, alpha-10, alpha-11, alpha-M, alpha-V, alpha-X, beta-1, beta-2, beta-4, and beta-5, some of which hold common to the recent study (Zhang et al., 2016).
Integrins play a vital role in providing structural integrity and attachment of cells with the extracellular matrix, facilitating signal transduction. These collagen-binding integrins can hence serve as potential therapeutic targets for eye diseases such as myopia (McBrien et al., 2006). The presence of such receptors in sclera substantiates further studies as their possible role in the biomechanical remodeling involved in ocular diseases.
Proteins uniquely identified in this study
The majority of proteins identified in our study have not been reported previously in the scleral proteome. To catalog the unique proteins from our study, we compared our results with the previous high-throughput study on the sclera (Zhang et al., 2016). In comparison, we observed that 2931 proteins were unique to our study, while 1524 proteins were common to both the studies. A partial list of these uniquely identified proteins is provided in Table 2, and the Venn diagram of the comparison is shown in Figure 2D. The corresponding spectra of four proteins uniquely identified in the sclera are provided in Figure 6A–D. The list includes radixin (RDX), synaptopodin (SYNPO), paladin (PALLD), brain abundant membrane attached signal protein 1 (BASP1), FYVE and coiled-coil domain containing 1 (FYCO1), netrin 1 (NTN1), and kelch-like family member 41 (KLHL41).
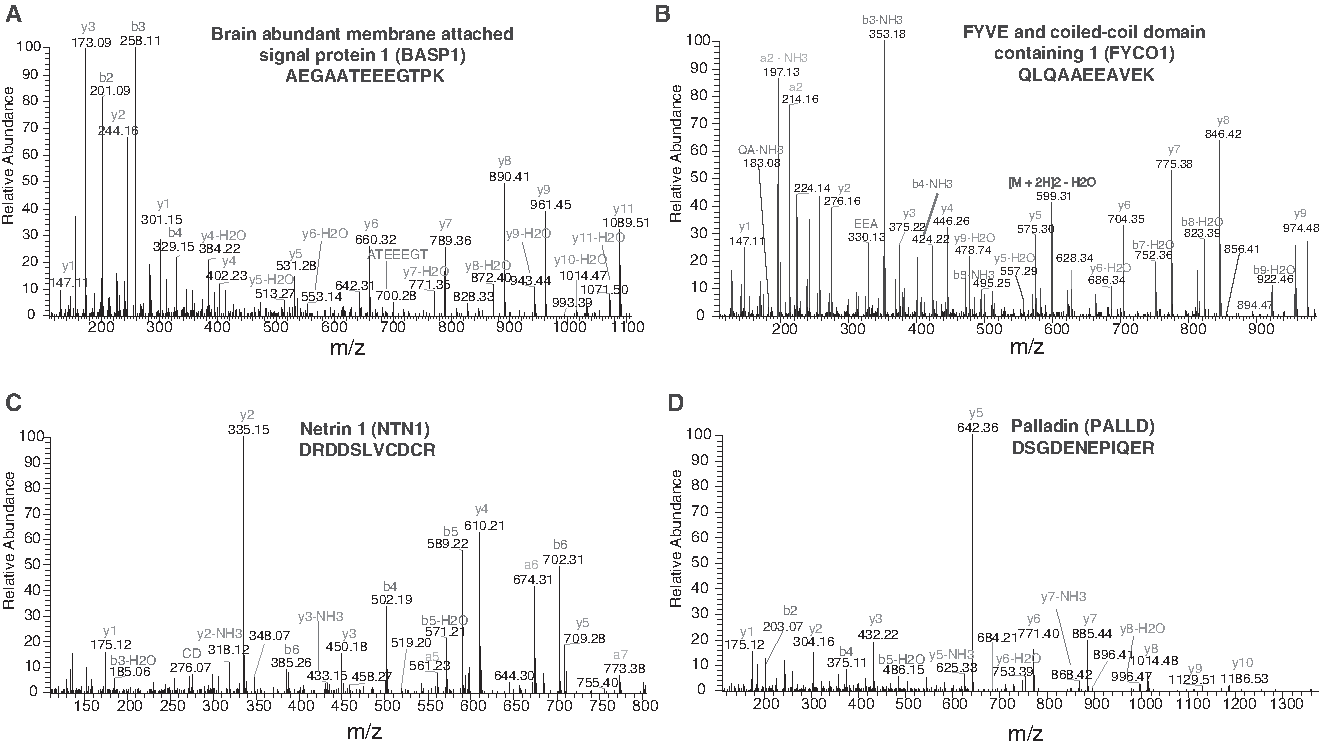
Representative MS/MS spectra of some proteins uniquely identified in this study.
A List of Unique Proteins Identified When Compared to a Previous High-Throughput Study (Zhang et al., 2016)
We have compared our list of proteins with a high-throughput study by Zhang et al. (2016) and observed 2931 proteins were unique to our study, while 1524 were common to both the studies.
RDX is a cytoskeletal protein encoded by the RDX gene and is a part of ezrin–radixin–moesin protein family. These scaffolding proteins play a significant role in cell motility and adhesion (Qin et al., 2014). Their localization to the plasma membrane allows transduction of signals between the extracellular matrix and the cytosol through different molecules such as CD95, CD44, CD43, and NHE1 (Denker et al., 2000; Lozupone et al., 2004; Tsukita and Yonemura, 1997; Yonemura et al., 1998). The viscoelastic properties and the presence of specialized projections such as filopodia and lamellipodia assist in cell shape maintenance (Lamb et al., 1997; Tachibana et al., 2015).
We also found other molecules such as SYNPO and PALLD, known to be involved in controlling the cell shape, adhesion, and contraction, suggesting their role in cell shape assembly and maintenance. There are also reports demonstrating the role of SYNPO in endothelial wound healing and cell–cell adhesion (Kannan and Tang, 2015; Mun et al., 2014). These findings suggest that along with RDX, SYNPO and PALLD may play a role in structural and functional dynamics in the sclera.
The immune system of the eye provides protection against the bacteria, viruses, and potential antigens, which is mainly done by conjunctiva, tear, and cornea. The sophisticated defense mechanism and the exclusive anatomical structure allow transudation into tissue upon infections and result in immune reactions. FYCO1 is known to contain a RUN domain and plays a significant role in the development of lens (Chen et al., 2011). The expression of FYCO1 has been previously reported in heart, skin, and ovary (Kiss et al., 2002). In 2011, Chen et al. carried out a genome-wide linkage analysis and identified that the mutation associated with this gene lead to congenital cataracts.
Studies are stating the involvement of FYCO1 in dectin-1 signaling, implicating its role in microbial killing by phagocytosis (Ma et al., 2014; Pankiv et al., 2010). Similarly, BASP1 is a membrane-bound protein with immunological characteristics. The localization of BASP1 has been initially reported in the nucleus, but later, the studies have demonstrated that the subcellular location depended highly on the two forms of the protein—myristoylated and nonmyristoylated (Carpenter et al., 2004; Ohsawa et al., 2008). Its main functions include regulation of synaptic growth and the actin cytoskeleton, thus suggesting its role in sclera development (Frey et al., 2000). Also, this protein has been detected in lens and cornea, but its role in these tissues has not been established, suggesting further studies for the characterization of BASP1 (Bagchi et al., 2008; Baudet et al., 2008).
NTN1 belongs to the laminin-related protein family consisting of axon guidance molecules. NTN1 is well known for its role in the development of the brain during embryogenesis (Graef et al., 2003; Serafini et al., 1994). It has a regulatory role in cellular motility, survival, and proangiogenic properties, suggesting its role in driving scleral vascularization (Sanvoranart et al., 2016; Shimizu et al., 2013). Some of its non-neuronal functions include angiogenesis and inflammation. However, the detailed molecular mechanisms are not known (Larrivee et al., 2007; Tadagavadi et al., 2010; Wilson et al., 2006).
Scleral proteins associated with autoimmune and ocular disorders
Inflammation of sclera is associated with specific autoimmune diseases (Akpek et al., 2004). It has been reported that ophthalmologic signs serve as early symptoms in 10% of systemic diseases where scleral association can be seen in 7–10% of patients (Galor and Thorne, 2007). Autoimmune disorders such as rheumatoid arthritis, Wegener Granulomatosis, Sjögren's syndrome, Systemic Lupus Erythematosus, and Graves' disease are often accompanied with detrimental systemic and ocular effects (Generali et al., 2015). Scleritis and episcleritis are the inflammatory diseases affecting sclera and its adjacent tissues. We identified a significant subset of proteins whose altered expression has been reported in many autoimmune and ocular disorders (Generali et al., 2015).
In this study, we identified proteins involved in inflammation such as complement proteins and S100 proteins. The S100 proteins are a family of calcium-binding proteins playing regulatory roles in numerous cellular processes such as such as cell cycle progression and differentiation, protection from oxidative cell damage, protein phosphorylation, and secretion (Santamaria-Kisiel et al., 2006).
We have identified several S100 proteins, such as S100A11, S100A16, S100A10, S100A13, S100A4, S100A8, S100B, S100A6, S100A7, S100A9, and S100A14. The altered expression of this family of proteins is linked with inflammatory diseases such as rheumatoid arthritis (Baillet et al., 2010; Bhattacharjee et al., 2016; Foell et al., 2004). Similarly, cystatin C, vascular cell adhesion molecule-1 and CXC chemokine ligand 4 identified in this study, is also known to be a potential marker for detecting systemic lupus erythematosus (Mok et al., 2018; Peixoto et al., 2015). Any abnormality in these proteins at an early stage will help in identifying the systemic diseases, which can further serve as therapeutic targets.
Conclusions
The results from this study provide a high-resolution mass spectrometry-based proteomic map of the human sclera. The current dataset serves as one of the most extensive catalog of scleral proteins. Our study also provides the functional analysis of 4493 nonredundant scleral tissue proteins where we observed enrichment of proteins in pathways such as cell adhesion, developmental process, and secretion, providing important insights to its function. This study also provides a valuable baseline for future investigations so as to map the dynamic changes that occur in sclera in various pathological conditions.
Footnotes
Acknowledgments
We thank Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka, for the support to the Center for Systems Biology and Molecular Medicine at Yenepoya (Deemed to be University) under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). We thank Yenepoya (Deemed to be University) for access to instrumentation. V.M. is a recipient of Junior Research Fellowship from Yenepoya (Deemed to be University). V.M. is a recipient of Women Scientist-A award from the Department of Science and Technology (DST), Government of India. M.A.N. is a recipient of Junior Research Fellowship from University Grants Commission (UGC), Government of India. S.K. is a recipient of Senior Research Fellowship from India Council of Medical Research (ICMR), Government of India. S.M.P. is a recipient of INSPIRE Faculty Award from DST, Government of India.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.