
Abstract

Pharmacovigilance and Medicinal Products
For the past century, modern medicines have positively contributed to public health and changed the ways human diseases are prevented and treated. Yet, most drugs are not without side effects. Adverse drug reactions (ADRs) rank among the leading causes of morbidity and mortality worldwide. The World Health Organization defines pharmacovigilance as “the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other possible drug-related problems” (WHO, 2006).
The scope of pharmacovigilance is not limited to ADRs. Other drug-related problems such as treatment failures, drug–drug and drug–food interactions, not to mention herbal, traditional, and complementary medicines, biologics, and vaccines are of concern to pharmacovigilance as well. Pharmacovigilance is broadly relevant for anyone and organization interested in safe and rational use of medicinal products.
The current paradigm of drug development can detect only the most common ADRs during premarketing clinical trials. One reason is that new drug candidates are tested in as few as 500 and rarely >5000 individuals in clinical trials (WHO, 2004). In addition, the range of disease characteristics and comorbidities evaluated in clinical trials is much narrower than those in real-life therapeutic contexts in the general population. Once a drug is set free for use outside the realm of controlled clinical trials, novel ADR signals begin to emerge as the diversity and size of the population exposed to the drug increase to many thousands and millions.
Could the existing pharmacovigilance paradigm be improved to detect a broader range of ADRs, including those that are uncommon, and well in advance of deploying new drugs in clinical practice?
Increasing the sample size in clinical trials would help identify more ADRs. But detecting pharmacovigilance signals on less common and rare ADRs requires large clinical trials that would face barriers in feasibility and timelines.
Rethinking how we recruit participants to clinical trials may offer, however, new insights on next-generation pharmacovigilance. Human subjects are recruited to clinical trials by attributes such as demographics, disease symptoms and severity, comorbidities, and prior treatment history, among other criteria. Although these criteria may partially relate to interindividual variability in drug pharmacokinetics (PK) and pharmacodynamics (PD), they do not offer direct mechanistic explanations on why drug exposure and performance differ vastly among patients and populations.
The current nonmechanistic approach to population sampling in clinical trials has an important corollary for safety and efficacy of medicinal products. Individuals who markedly differ from population averages in PK and molecular target variation are exposed to new therapeutic candidates and tested for safety and efficacy much later in the clinical trial recruitment process, and more often than not, long after drugs are already introduced for routine use in the general population.
For example, consider a case of drug toxicity associated with a poor drug metabolism trait that has a prevalence of <0.1% in the population. Most clinical trials would be statistically underpowered to detect such low-frequency ADRs. The timing of pharmacovigilance signal detection would shift from clinical trials to routine clinical practice when the size of the drug-exposed population is sufficiently large to detect weak ADR signals.
Next-Generation Pharmacovigilance
Clinical trial design by extremely discordant biomarkers
Precision medicine enthusiasts tend to refer to the current era of pharmacology and toxicology as the golden age of biomarkers. Although molecular studies of PK and PD variability have been conducted for decades, routine use of biomarkers in drug development and clinical practice is relatively recent (Alberts, 2012; Biomarkers Definitions Working Group, 2001; Kalow et al., 1999). Biomarker applications are not limited, however, to precision medicine or understanding PK and PD variability at a resolution of individuals. Biomarkers can also help establish the upper and lower bound limits of PK and PD variability in a given population as well as between populations.
For example, cytochrome P450 2D6 (CYP2D6) is one of the most intensively researched drug metabolism pathways. CYP2D6 displays marked genetic variability in its catalytic function that differs by several orders of magnitude person-to-person and across populations. Individuals can be functionally assigned as CYP2D6 poor or ultrarapid metabolizers as determined by genetic and/or phenotypic biomarkers. Hence, one can infer on population upper and lower bound limits of drug exposure for therapeutics that are primarily cleared by CYP2D6. In the case of functional variations in drug targets, they can cause changes in potency (EC50), efficacy (Emax), or both. Functional variation in many drug metabolism pathways and molecular drug targets has already been mapped across populations worldwide.
The lesson for our purposes is that many PK and PD biomarkers already exist whose known variations in the general population can be utilized to estimate the upper and lower bound limits for variability in drug exposure and molecular targets. By mechanistic recruiting in clinical trials, we can proactively include the patients and healthy volunteers who display biomarkers that confer PK and PD phenotypes situated in population edges (e.g., ultrarapid or poor drug metabolism, or those with very high or low EC50 and Emax values). In such contrasting panels of patients or healthy volunteers representing the population extremes for PK, PD, and molecular targets, we can estimate upper and lower bound limits for drug safety and efficacy in a given population.
Once drug safety and efficacy are evaluated under such extreme (population edge) conditions with respect to drug exposure and molecular target variation, clinical trial outcomes of subjects with less extreme PK and molecular target traits become more foreseeable, that is, residing within our estimates for population upper and lower bound limits of drug-related traits.
As with aircrafts or automobiles tested in wind tunnel experiments, our main premise is that drugs, too, warrant “stress testing” proactively so as to identify, early on, their performance under conditions that mimic the population extremes (edges) in regard to distribution of PK traits and molecular drug targets.
By contrast, the current traditional approach to pharmacovigilance signal detection is much slower. The inclusion of persons in population edges is left to the incremental and slow increase in the number of the drug-exposed population sample by nonmechanistic recruitment of subjects.
We named the proposed proactive and a priori panel testing of patients or healthy volunteers with extremely discordant biomarker (EDB) traits (e.g., ultrarapid or very slow drug metabolism) as the “EDB clinical trial design” (Fig. 1). If this newer trial design were to become an integral part of the phase 1 or phase 2 clinical trials in drug development, it could possibly help overcome some of the existing pharmacovigilance bottlenecks, potentially accelerating pharmaceutical innovation.
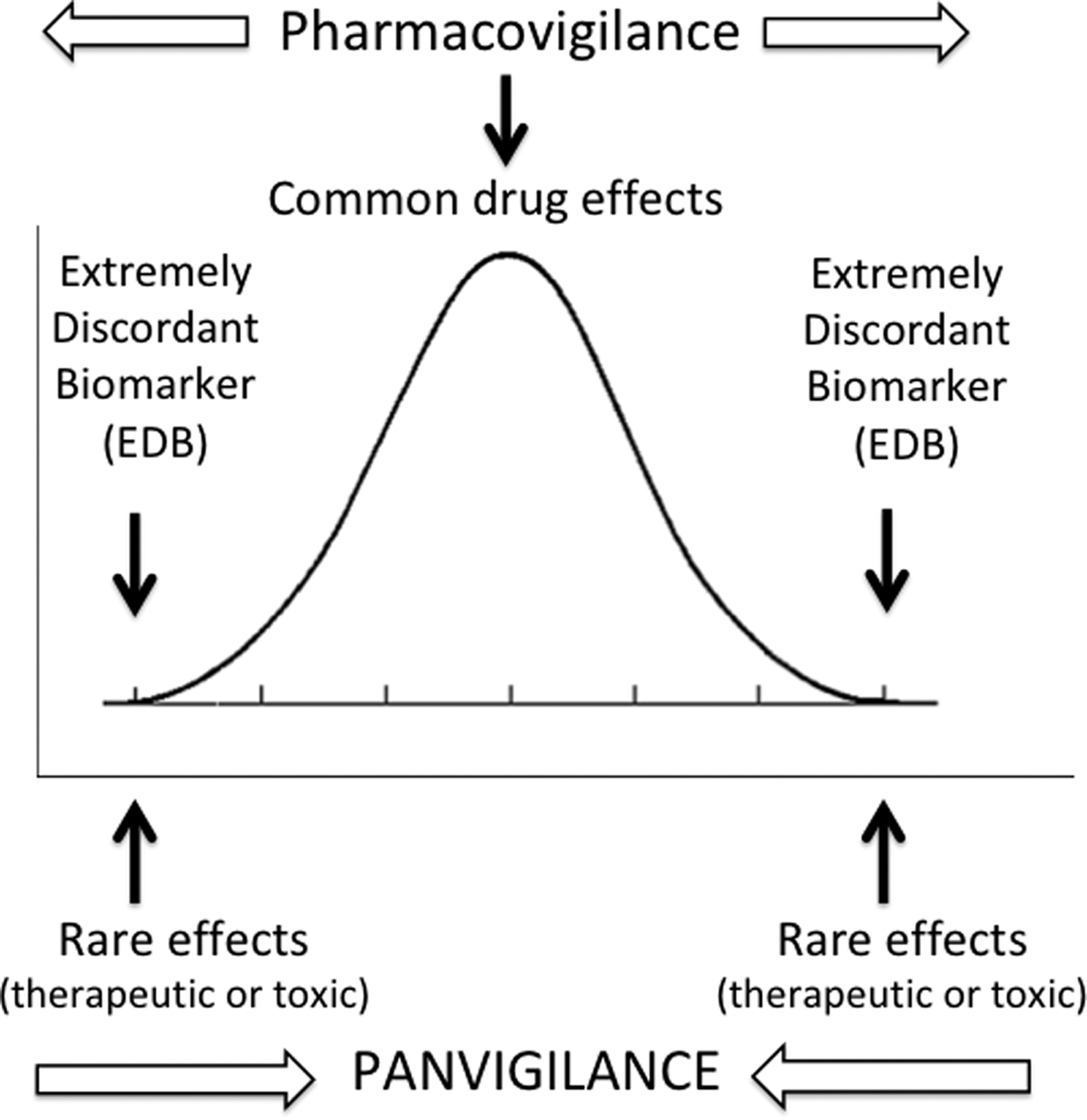
Comparison of panvigilance with traditional pharmacovigilance. Panvigilance is based on forecasting of signals on unknown drug effects, be they adverse, toxic, or therapeutic, by prioritizing PK and PD analyses in population edges as defined by biomarkers. Note that the direction of discovery for drug PK and PD effects is “from rare and less common to common” in the case of panvigilance. This contrasts with traditional pharmacovigilance that detects, first, common drug effects in controlled clinical trials, followed by less common and rare drug effects in the course of routine clinical use as drug exposure increases in the general population. PD, pharmacodynamics; PK, pharmacokinetics.
Ideal Timing for EDB-Guided Clinical Trials?
Because the panels of patients or healthy volunteers harbor the PK and PD biomarkers in population edges, caution should be exercised to prevent drug toxicity or treatment failures in the course of signal detection in such pharmacovigilance clinical trials. Single-dose safety and efficacy studies in persons with EDBs would be prudent whenever possible, rather than multiple dose studies. As an additional intent of pharmacovigilance studies with EDBs is to inform large-scale confirmatory clinical trials (e.g., in phase 3 drug development and afterward), phase 1 and/or phase 2 clinical trials in early drug development would bode well for the proposed EDB-guided pharmacovigilance clinical trial design. In common diseases such as hypertension where PD endpoints or surrogates also exist in healthy volunteers (e.g., blood pressure), the upper and lower bound limits for molecular target variations can be tested in single-dose studies in healthy volunteers as well.
Outlook on Panvigilance and Systems Pharmacovigilance
Origins of the drug industry date to the 19th century when organic chemicals derived from distillation of coal (coal-tar) in the textile and synthetic dye industries have paved the way for the first pharmaceutical companies. Although the innovation bottlenecks in drug development more than a century ago were in part related to access to raw materials, the current bottlenecks are much different in the pharmaceutical industry. Automation and extreme digital connectivity (Özdemir, 2018a, 2019a; Read, 2018), knowledge governance (Özdemir, 2018b, 2019b; Ravetz, 2016; Rip, 2016; Sarewitz, 2016; Stilgoe et al., 2014; Thoreau and Delvenne, 2012; von Schomberg, 2013), “variability science” or the art and science of forecasting, and governing person-to-person and population scale variations in drug PK and PD traits are some of the modern-day bottlenecks in pharmaceutical innovation (Kalow et al., 1999, 2001; Özdemir et al., 2005).
We introduced and described in this article two related new concepts, panvigilance and EDB-guided clinical trials, with a view to next generation strategic risk governance for medicinal product development and regulation. The proposed clinical trial design for panel testing of patients and healthy volunteers with EDBs would usefully inform early phase drug development, dose finding in phase 3 clinical trials, and boost pharmacovigilance signal detection. Biomarker discoveries have been aided by the concept of extreme discordant phenotypes over the past two decades (Nebert 2000). In 2019, we have now access to a large inventory of biomarkers already discovered and developed that can be used as EDBs and thus, broaden the field of pharmacovigilance to systems pharmacovigilance and panvigilance.
Clinical trials designed with EDBs would transform pharmacovigilance to panvigilance as the entire complement of PK and PD variability and their upper and lower bound limits can be more effectively captured. Needless to say, traditional pharmacovigilance and the panvigilance approach for full-scale PK and PD variability assessment are not mutually exclusive and complement each other for safe and rational development and use of medicinal products (Fig. 1).
By virtue of mechanistic sampling in clinical trials using EDBs, panvigilance offers the advantage to “road test” a drug candidate under extreme PK and PD conditions before introduction for clinical and population use. We suggest that because drugs impact public health greatly, drug discovery and development should have no lesser standards for road testing of new therapeutic candidates than aircraft design and development.
We cannot eliminate variability in drug PK and PD, but we can certainly govern it more effectively, and with strategic foresight gained from clinical trials that incorporate EDBs.
Footnotes
Disclaimer
Views expressed are authors' personal opinions only and do not necessarily reflect those of the affiliated institutions.
Acknowledgements
No funding was received in support of this article. The concept of panvigilance was first presented by V.Ö. on November 6, 2015 at the Pharmacovigilance Congress in Ankara, Turkey.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.