
Abstract
Congenital heart diseases (CHDs) are complex traits that manifest in diverse clinical phenotypes such as the Tetralogy of Fallot (TOF), valvular and ventricular/atrial septal defects. Genetic mechanisms of CHDs have remained largely unclear to date. Copy number variations (CNVs) have been implicated in many complex diseases but their impact has not been examined extensively in various forms of CHD lesions. We report in this study, to the best of our knowledge, the largest cohort of Saudi Arab CHD patients to date who were evaluated using genome-wide CNV analysis. In a sample of 134 Saudi Arab patients with CHD, 66 exhibited pathogenic or likely pathogenic CNVs. Notably, 21 copy number gains and 11 copy number losses were detected that encompassed 141 genes and 146 genes, respectively. The most frequent gains were on 17q21.31, 8p11.21, and 22q11.23, whereas the losses were primarily localized to 16p11.2. Interestingly, all lesions have had gains at 17q21.31. Septal defects had also gains at 8p11.21 and 22q11.23, valvular lesions at 8p11.21, 22q11.23, and 2q13, and TOF at 16p11.2. Functional and network analyses demonstrated that cardiovascular and nervous system development and function as well as cell death/survival were most significantly associated with CNVs, thus highlighting the potentially important genes likely to be involved in CHD, including NPHP1, PLCB1, KANSL1, and NR3C1. In conclusion, this genome-wide analysis identifies a high frequency of CNVs mostly in patients with septal defects, primarily influencing cardiovascular developmental and functional pathways, thereby offering a deeper insight into the complex networks involved in CHD pathogenesis.
Introduction
Congenital heart diseases (CHDs) constitute a developmental malformation of the heart, aorta, or other large blood vessels that make up the most common forms of major birth defects in newborns (Tennant et al., 2010). Clinically, these defects present in different forms, such as atrial septal defects (ASDs), ventricular septal defects (VSDs), pulmonary, mitral/aortic valvular stenosis/incompetence, coarctation of the aorta, Tetralogy of Fallot (TOF), transposition of the great arteries (TGA), occurring as single defects or in combination with other developmental anomalies or syndromes.
Although it is widely acknowledged that CHDs are highly heritable in Mendelian, polygenic, or sporadic forms, their precise etiology remains largely unknown. Thus, while rare familial monogenic forms have been described, these defects are likely to result from heterogeneous and multiple types of genetic alterations, including point mutations or variants in genes encoding cardiac structural proteins and genomic instability (An et al., 2016; Bansal et al., 2014; Costain et al., 2016a; Glessner et al., 2014; Goldmuntz et al., 2011; Hitz et al., 2012; Ji et al., 2019; Wessels and Willems, 2010).
Understandably, different study platforms, such as genome-wide association studies (GWASs), exome sequencing, targeted gene approaches, and molecular karyotyping have yielded varying results with respect to genetic causes and the disease pathways leading to CHD. This points to the probability of these lesions underlying various, partly converging mechanisms involved in developmental disease pathways. Nonetheless, these lesions are, by and large, presumably a product of an abnormal dosage of one or more gene(s) within chromosomal regions, resulting from gains or losses in genomic copy numbers (Al-Hassnan et al., 2018; Costain et al., 2016b; Greenway et al., 2009; Soemedi et al., 2012; Thomford et al., 2018).
While copy number losses (CNLs) lead to a straightforward effect on the genomic sequence, copy number gains (CNGs) can trigger very diverse effects on gene function, depending on the breakpoints, location, or gene reading frame (Conrad et al., 2010; Deak et al., 2011; Newman et al., 2015). Thus, chromosomal duplications can cause certain severe phenotypes through loss or gain of function, triplosensitivity, and/or dysregulation of genes within or near the duplicated region (Newman et al., 2015). The impact of such variations on CHD manifestation is not fully explained yet.
Additionally, although it is now well acknowledged that deletions or duplication may be responsible for clearly defined CHD phenotypes, it is not certain whether these disorders are discernible at the level of delineating loci for the causative copy number variations (CNVs). We hypothesize that, being predominantly cardiac developmental disorders, polygenic diseases such as CHDs are likely to involve common genes engaged in cardiovascular developmental pathways as well as sharing gene/gene interaction systems involved in cardiac structural formation and compartmentalization.
We were therefore interested in evaluating the impact of genomic imbalances as well as related functional pathways and network systems on the manifestation of these disorders employing the Saudi Arab population as a study model. Importantly also, there is a rapid inclination toward utilizing the knowledge base of these CNVs for clinical purposes (Al-Hassnan et al., 2018; Geng et al., 2014; Hanchard et al., 2017; Kim et al., 2016; Monteiro et al., 2017; Southard et al., 2012), rendering it conducive to evaluate the importance of such genetic variations in any given population with a large candidate base for CHDs such as Saudi Arabia.
In the present study, we evaluated the likelihood of the different CHDs displaying unique patterns ascribable to particular forms of genomic instability, in search of potential tools for clinical genetic diagnostic purposes. This was accomplished by performing whole-genome CNV analyses in 134 Saudi Arab patients who have various forms of CHD lesions. The findings offer original molecular leads and insights while also confirming some of the candidate genes reported previously in the literature at the implicated genomic loci.
Materials and Methods
Patient recruitment and phenotypic classification
The initial study cohort consisted of 140 Saudi Arab individuals who underwent surgical repairs for a variety of CHD lesions at our institution. Clinical data were collected from patient histories and chart reviews. Patients identified as harboring Down and Kartagener's syndromes were excluded from the final analysis. The final cohort consisted of 134 unrelated patients (Table 1 and detailed clinical and phenotypic characteristics are given in Supplementary Table S1), who were classified as simple/single lesions (ASDs and VSDs), denoted as Group 1, valvular heart disease (mitral valve disease, pulmonary valve disease, aortic valve disease, and tricuspid valve disease) as Group 2, aortic arch disease as Group 3, TOF as Group 4, and all other complex CHD as Group 5 (Supplementary Table S1). The Venn diagram approach was employed for identifying CNVs among and specific to each group.
Clinical and Phenotypic Characteristics of All Patients in the Study (N = 134)
Atrial septal defect and ventricular septal defect.
Mitral valve disease, pulmonary valve disease, aortic valve disease, and tricuspid valve disease.
BMI, body mass index; CHD, congenital heart disease.
The study was approved by the Institutional Review Board (IRB) of the King Faisal Specialist Hospital and Research Center (KFSHRC) and performed in accordance with the current version of the Helsinki Declaration. All participants signed written and informed consent.
CNVs by genome-wide single nucleotide polymorphism arrays
The Affymetrix whole genome chromosome single nucleotide polymorphism (SNP) microarray was employed to assess for genomic imbalances (i.e., gain or losses) using the Genome-Wide SNP Array CytoScan HD. This platform contains 2.6 million markers for Copy Number Variant detection (Affymetrix, Inc.) covering the whole genome, comprising 750,000 genotype SNPs and 1.9 million nonpolymorphic probes. Data were analyzed using the Affymetrix® Chromosome Analysis Suite (ChAS), version Cyto 2.0.0.195 (r5758). Oligonucleotide probe information was based on the 37 build of the Human Genome (UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway, hg19).
Briefly, 250 ng of genomic DNA was digested with NspI, ligated to an adapter and amplified by polymerase chain reaction (PCR) using a single pair of primers that recognized the adapter sequence. The PCR products were run on a 2% TBE gel to confirm the size of the majority of products between 150 and 2000 bp in length.
To procure adequate PCR material, all products were combined for each sample and purified using magnetic beads (Agencourt AMPure; Beckman Coulter, Beverly, MA, USA). The purified PCR products were fragmented by DNase I and visualized on a 4% TBE agarose gel to attain fragment sizes ranging from 25 to 125 bp. The fragmented PCR products were subsequently end-labeled with biotin, hybridized to the array, washed, stained with a GeneChip® Fluidics Station 450, and scanned on the Affymetrix GeneChip Scanner 3000 7G. Scanned data files were generated using Affymetrix GeneChip Command Console Software, version 1.2, and analyzed with ChAS v3.1 (Affymetrix, Inc.).
The Hidden Markov Model (HMM) within the ChAS software package was used to determine the copy number types and their breakpoints, employing thresholds of log2 ratio ≥0.58 and ≤−1 to categorize altered regions as CNGs (amplification) and CNLs (deletions), respectively. To avoid false-positive CNV detection due to inherent microarray “noise,” only alterations involving at least 50 consecutive probes and more than 200 kb in size were considered in classifying the altered regions. The detected CNVs in our patients were then evaluated based on the American College of Medical Genetics and Genomics standards and guidelines (South et al., 2013). The aberrations representing common benign CNVs were excluded by comparing all the identified CNVs with those reported in the Database of Genomic Variants (http://projects.tcag.ca/variation/) and in our own database for individuals who have been classified as normal.
Functional pathway and network analysis
Gene ontology enrichment, functional, pathway, and interaction network analyses of copy number variant loci genes were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) (Sherman et al., 2007) and QIAGEN Ingenuity Pathway Analysis (IPA®; QIAGEN, Redwood City, CA, USA). IPA provides the ability to analyze and identify altered pathways, as well as detecting interactions among multiple genes, and building their interaction networks. Using a 99% confidence level, scores of ≥2 were considered significant. A right-tailed Fisher's exact test was employed to calculate the p-value for the probability of the biological function (or pathway) assigned to that data set being explained by chance alone.
Replication of CNV by quantitative PCR
We randomly selected six of the studied individuals based on the CytoScan HD findings of the CNGs and CNLs at six randomly selected loci to confirm the array findings by real-time PCR using the ABI Prism 7900HT sequence detection system (Applied Biosystems, Warrington, United Kingdom) (Supplementary Table S2). We used TaqMan copy number probes sourced from Applied Biosystems. These probes were tagged with FAM and VIC fluorescent dyes to detect the target gene (FAM dye) and a reference sequence known to exist in two copies in a diploid genome.
Briefly, for each reaction, 1.0 μL each of test and calibrator CNV probes were mixed with 40 ng DNA in 4 L, 10 μL of 2 × TaqMan genotyping master mix, and 4.0 μL of nuclease-free water, in a total volume of 20 μL as per the manufacturer's protocol. The mixture was dispensed in four replicates into a 96-well plate and three no-template controls were set up for each reaction. The plate was subjected to the following PCR program: 10 min at 95°C to prime the enzyme, and 40 cycles of 95°C for 15 sec and 60°C for 60 sec. Data were analyzed using the Copy Caller software V2.0 (Applied Biosystems, Carlsbad, CA, USA).
Results
Detection of CNVs in CHD patients
The 134 patient samples were subjected to the CNV analyses (Table 1) that resulted in 66 individuals displaying positive and 68 with negative arrays (patient details in Table S1). The affected individuals included 29 atrial and septal defects; 14 aortic, mitral, tricuspid, and pulmonary valve diseases; 10 aortic arch cases; 8 Tetralogy of Fallot (TOFs), and 5 complex congenital heart disorders (Fig. 1 and Supplementary Table S3). In all, 32 CNVs were detected among the affected patients, with a median size of 571.8 kbp (min 100.1 kbp, max 4.6 Mbp) (Table 2 and Fig. 1), whereby 21 gains encompassed 141 genes and 11 losses affected 146 genes (Table 3). The most frequent gains among all patients were localized to chromosome (chr) 17q21.31, 8p11.21, and 22q11.23, whereas the losses were described on 16p11.2 (Fig. 2).

Distribution of CNVs across patients from five different congenital cardiac lesions. Different congenital cardiac lesion groups are color coded. Gain is shown in blue and loss is in red. CNV, copy number variation.
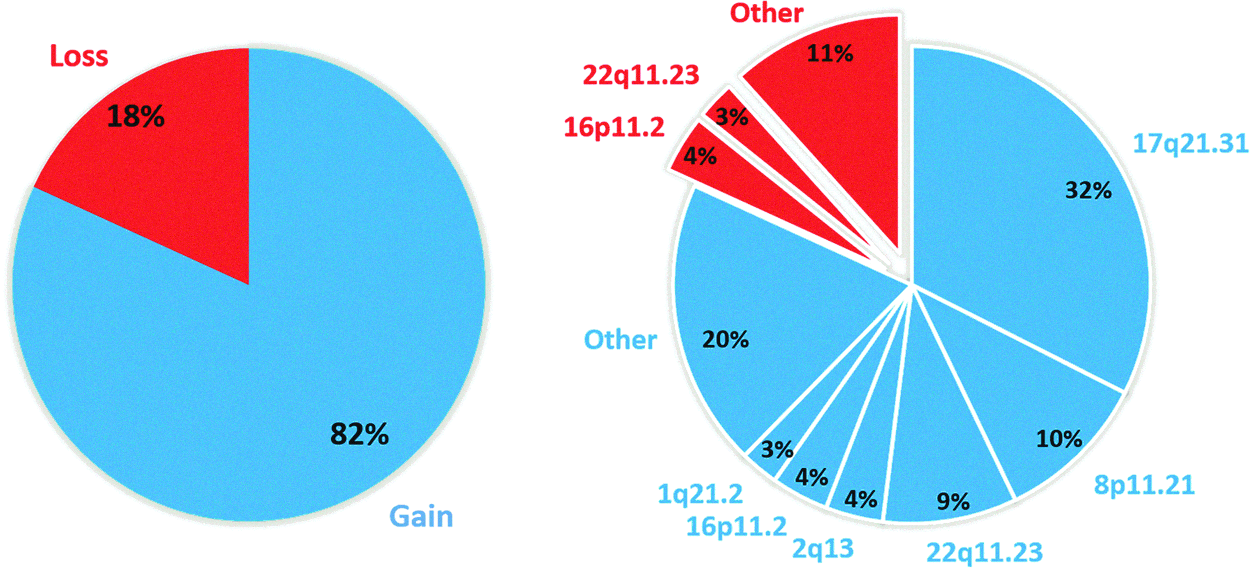
Distribution of CNVs among all patients. Blue and red indicate CNG and CNL, respectively. CNG, copy number gain; CNL, copy number loss.
The List of Genes Located in Copy Number Variation Loci
List of Copy Number Variations in Five Different Types of Congenital Cardiac Lesions
CNV, copy number variation; Group 1, simple/single lesions (atrial septal defect and ventricular septal defect); Group 2, valvular heart disease (mitral valve disease, pulmonary valve disease, aortic valve disease, and tricuspid valve disease); Group 3, aortic arch disease; Group 4, Tetralogy of Fallot; Group 5, all other complex congenital heart diseases.
Accordingly, 32% of the pathogenic CNVs were primarily on chr 17q21.31, which harbors the KANSL1 gene locus, 10% on chr 8p11.21, 9% on chr 22q11.21, 4% on chr 16p11.2, 4% on chr2q13 at the nephronophthisis 1 (NPHP1) gene locus, 3% on 1q21.2, as well as 20% on other single loci. Classified by lesion, among all groups, CNVs were most recurrent on 17q21.31 that similarly harbored the highest portion of all identified variations. The CNVs on 17q21.31 have been previously shown to cause Koolen-de Vries syndrome (MIM: 610443) (Koolen et al., 2006). Figure 3 illustrates the distribution (percentages) of CNGs among patients in each group.
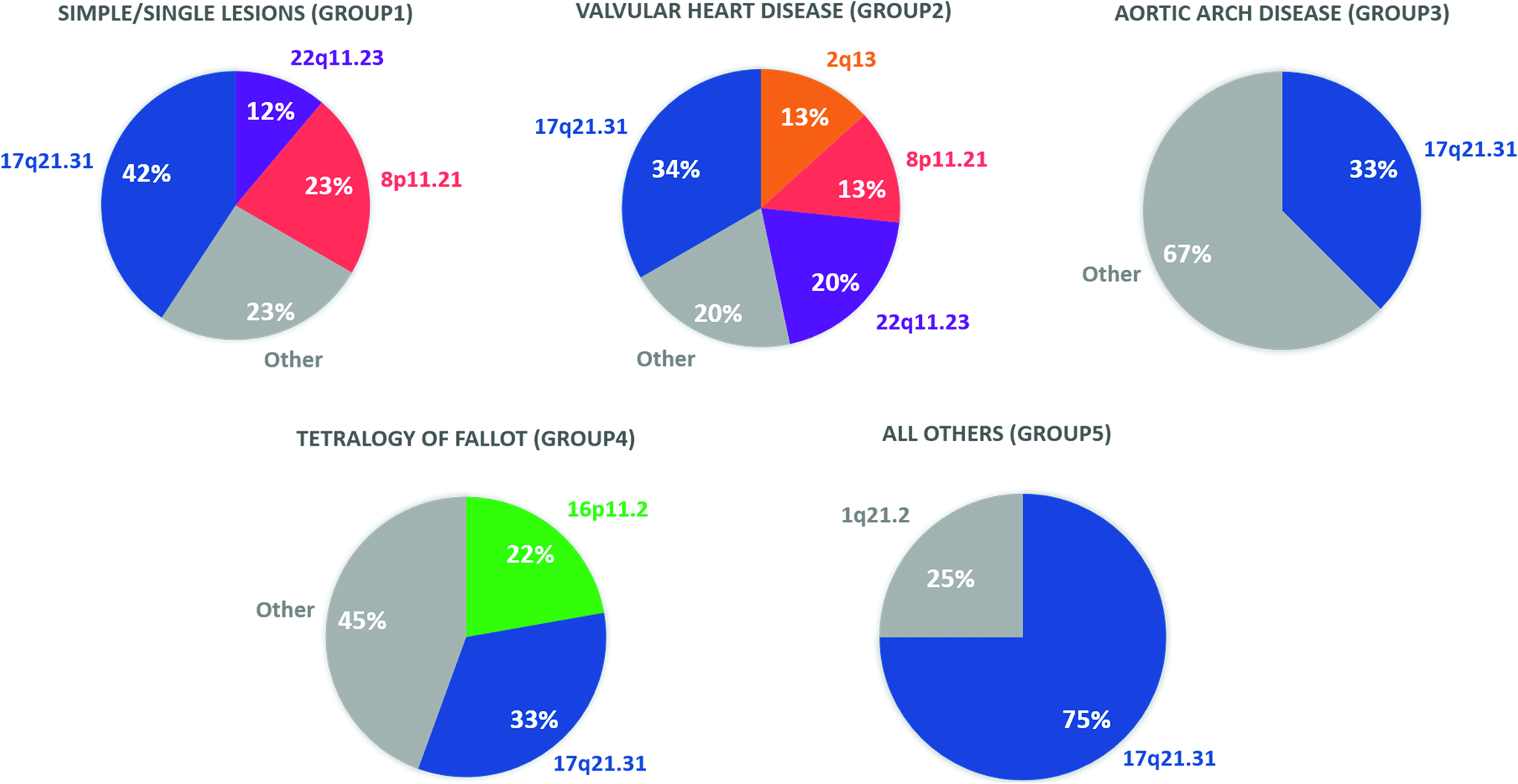
Pie charts representing the distribution of CNGs among all patients in each congenital cardiac lesion group. There were 26, 15, 9, 9, and 4 CNGs in Group 1 to 5, respectively.
Accordingly, septal defects were mainly associated with CNGs at 17q21.31 (42%), 8p11.21(23%), and 22q11.23 (12%); valvular lesions with gains at 17q21.31 (34%), 8p11.21 (13%), 2q13 (13%), and 22q11.23 (20%); the aortic arch disease with 17q21.31 (33%); and TOF with 17q21.31 (33%) and 16q11.2(22%), whereas 75% of all other lesions were linked to 17q21.31. Notably, quadruple CNGs on the 8p11.21 and 22q11.23 were linked to septal defects, which were specifically localized to the LRP5L and CRYBB2P1 on the 22q11.23, whereas the gains at the NPHP1 locus on 2q13 were associated with valvular lesions, aortic arch disease, as well as TOF. Additionally, several of the other individual loci were also singularly linked to various CHDs. These include the gain at 1q21.2 that was implicated in TOF.
Common and specific CNVs among different congenital cardiac lesions
Specific or common CNVs among five different congenital cardiac lesion groups were obtained using Venn diagram approach (Fig. 4A). Accordingly, CNG at 17q21.31, encompassing KANSL1, KANSL1-AS1, LRRC37A, ARL17A, ARL17B, NSFP1, LRRC37A2, and NSF, was shared by all CHD lesions. This CNV involved several genes that may play an important role in CHD, including KANSL1, which was linked to a quadruple gain in TOF in the present study. Besides, six CNGs and CNLs each were specifically ascribable to the simple/single lesion group (ASDs and VSDs), whereas seven gains and three losses were linked to the aortic arch disease (Fig. 4A, B). For valvular heart diseases, we detected seven CNGs with three, 5p15.2, 11q13.5, and 16p13.3, which were specific to this group only.
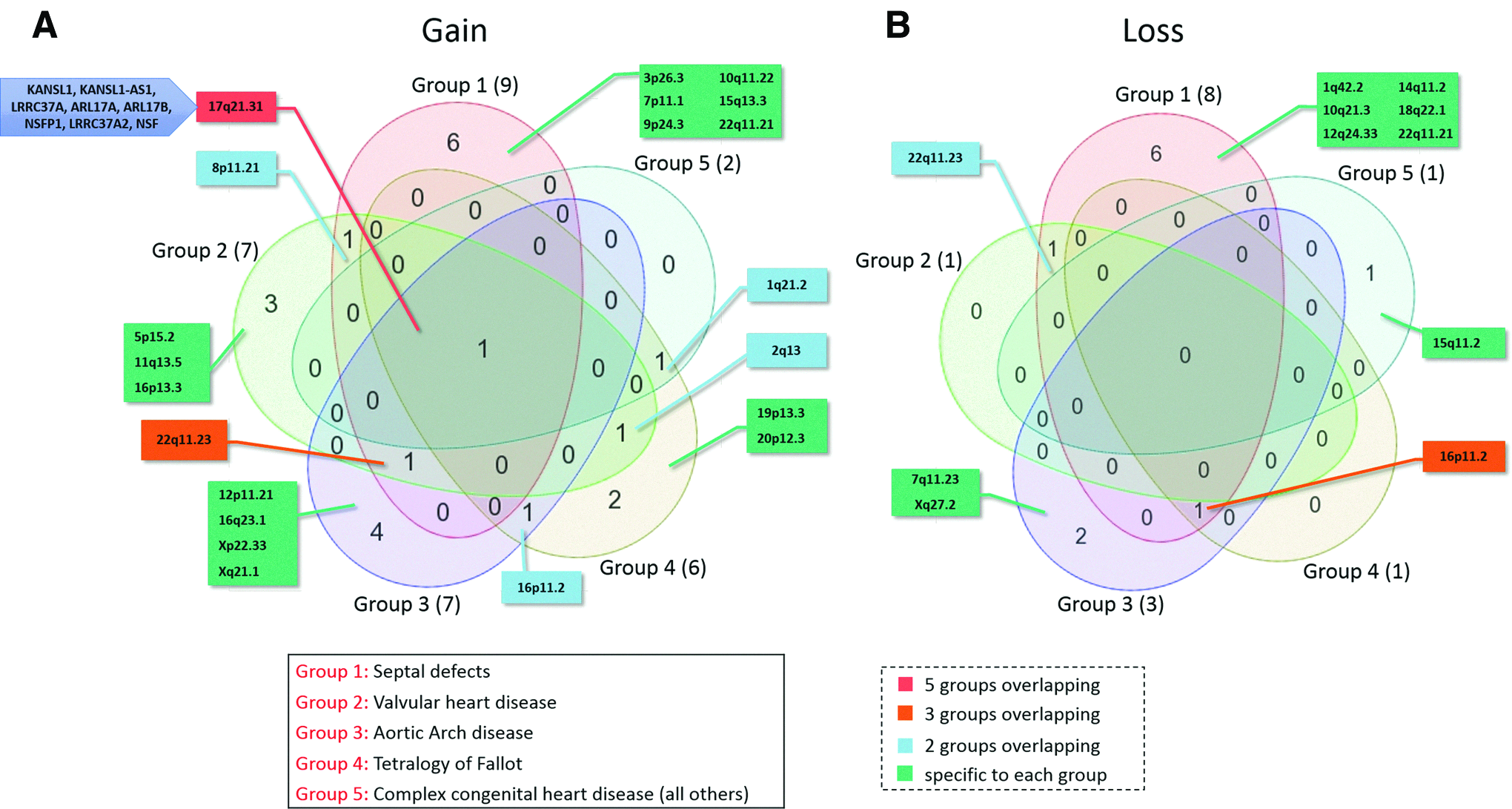
Venn diagram representing the identified CNGs
Gene ontology enrichment, functional, and network analyses
Gene ontology enrichment analyses were performed on all genes contained within the identified CNV loci (listed in Table 3) using DAVID to examine the annotated biological processes, canonical pathways, and diseases impacted by CNVs in CHD patients. Accordingly, the 10 most significantly enriched biological processes included protein peptidyl-prolyl isomerization, cell/cell adhesion, vesicle-mediated transport, cognition, endocytosis, phagocytosis, cell adhesion, wound healing, blood coagulation, and ribonucleoprotein complex biogenesis.
To obtain deeper insight into the gene network interactions, associated diseases and functions of the CNV loci genes, we performed functional and network analyses (Al-Harazi et al., 2019). The IPA analysis revealed that genes within CNGs are mainly involved in organismal injury and abnormalities, cell-to-cell signaling and interaction, molecular transport, cellular assembly and organization, tissue development, and cardiovascular disease (Fig. 5A), whereas the most enriched diseases and functions in CNLs were cardiovascular disease, drug metabolism, lipid metabolism, cell-to-cell signaling and interaction, and organismal development (Fig. 5B).

Gene ontology and functional analyses of genes located in CNVs. X-axis indicates the significance [–log10 (p-value)] of the functional/pathway association that is dependent on the number of genes in a class as well as biological relevance. The threshold line represents a p value of 0.05.
Furthermore, the most evidently altered canonical pathways for genes within CNGs include a neuroprotective role of THOP1 in Alzheimer's disease, amyotrophic lateral sclerosis signaling, proline degradation, nNOS signaling in neurons, and opioid signaling pathway (Fig. 5C). Among the losses, the top significant canonical pathways were the opioid signaling pathway, leukocyte extravasation signaling, L-DOPA degradation, and signaling by Rho family GTPases, and RhoGDI (Fig. 5D).
Gene interaction network analysis of CNV loci genes highlighted potentially the most essential genes likely to be involved in CHD, including NPHP1, PLCB1, and NR3C1, as indicated by the top subnetwork that is significantly enriched with genes related to cardiovascular system development and function, cellular development, cellular growth and proliferation, and nervous system development and function (Fig. 6A). In addition, network of interaction of CNL loci genes indicated a significant subnetwork that is related to cell death and survival, cardiovascular system development and function, and inflammatory disease (Fig. 6B).

The significant subnetworks of genes located in CNVs. Colored nodes are genes from the input list of genes in CNVs and uncolored ones are the interactors. Straight and dashed lines represent direct or indirect interactions, respectively. Genes in CNG are shown in blue
Validation of the SNP array results by quantitative PCR
Several CNVs were observed within multiple gene loci of potential interest with respect to CHD manifestation. These include the gains on chr 17q21.31 linked to several genes, including KANSL1, on 2q13 encompassing the NPHP1, on chr 1p31.36 harboring the NPHP4 gene, at 20p12.3 within the PLCB1 gene, on chr 19p13.1 carrying the ELANE gene, as well as the deletion at 15q11.2 within the region of the NIPA1 gene sequence. Our qPCR verification assays enabled us to replicate the array findings of CNG in the KANSL1 genes on chr 17q21.31, NPHP1 gene loci on chr 2q13, the ELANE gene on chr 19p13.3, the PLCB1 gene on chr 20p12.3, as well as the deletion within the NIPA1 gene sequence on chr 15q11.2 (Supplementary Table S2).
Discussion
We report in this study, to the best of our knowledge, the largest cohort of Saudi Arab CHD patients to date who were evaluated using genome-wide CNV analysis. In the present sample of 134 Saudi Arab patients with CHD, 66 exhibited pathogenic or likely pathogenic CNVs. Notably, 21 CNGs and 11 CNLs were detected that encompassed 141 genes and 146 genes, respectively.
Our study provides new insights into the impact of genomic instability and related disease pathways contributing to various forms of CHD lesions by using genome-wide high-density SNP arrays. Our results revealed a high yield of CNVs associated with various types of CHDs in the Saudi population. These changes stretched throughout the genome on different chromosomal loci, in the form of duplicate, triplicate, or quadruple gains, as well as deletions. They were linked partly to various genes at these loci, including for example ELANE, KANSL1, NIPA1, NPHP1, and PLCB1, which we were able to replicate. Noteworthy is the observation that by far the greatest majority of CNGs were linked to various forms of CHDs on chr 17q21.31. These disorders range from simple septal and valvular defects to complex disorders involving TOF, bicuspid aortic valve, and subaortic membrane disorder, among others.
Notably, one CNG at this locus was shared by all groups of lesions, possibly pointing to a common underlying trigger for these disorders in this genomic region. This region harbors several genes of potential interest, including KANSL1, KANSL1-AS1, LRRC37A, ARL17A, LRRC37A2, NSFP1, and NSF (Fig. 1). Of these, KANSL1 was linked to a quadruple gain in TOF. This gene encodes a nuclear protein constituting a subunit of two protein complexes, the MLL1 and NSL1, believed to be involved in histone acetylation in transcriptional regulation. This observation calls for further investigation on the precise function of this gene and its role in CHD.
The altered disease and functional categories implicated in CNGs in the present study were most significantly related to DNA replication, recombinant and repair, transcription regulator networks, protein peptidyl-prolyl isomerization, and intra-Golgi vesicle-mediated transport regulation of cell shape, to name a few. Since perturbations in the DNA synthesis processes are likely to induce an increase in gene copy numbers, our observations point to genomic instability, transcription, and regulation being deeply engaged in pathways leading to a variety of these disorders.
Also, given that the CNVs affected primarily cardiovascular developmental and disease pathways as well as cardiac-related gene/gene interaction systems, and considering the enrichment of structural lesions and signaling processes likely to be affected by these genomic changes at the 17q21.31, it can be speculated that this locus hosts essential genes regulating cardiac chamber formation and compartmentalization.
Apart from the chr 17q21.31, two other loci exhibiting eight gains each, the 8p11.21 and 22q11.23, were also implicated in several disorders (Ben-Shachar et al., 2008; Puvabanditsin et al., 2019). To begin with, the 8p11.21 harbors a quadruple gain in CHD. This locus contains, among others, the FNTA, SGK196, HGSNAT, and POTEA genes, which seem not to have been linked to CHD to date. Furthermore, the 22q11.23 carries the IGLL3P, LRP5L, CRYBB2P1, and ADRBK2. Interestingly, in the present study, the quadruple gain linked to septal defects on this site was localized to the LRP5L and CRYBB2P1 positions, whose cardiovascular or developmental functions have yet to be clearly defined.
Genomic sequences displaying quadruple CNGs also included the 20p12.3, which harbors the PLCB1 gene. This gene encodes enzymes involved in the formation of 1,4,5-triphosphatase and diacylglycerol from phosphatidylinositol 4,5-bisphosphatase that mediates the intracellular transduction of many extracellular signals. It resides within the genomic sequence stretch displaying both duplications and deletions implicated in complex disorders, in general, and associated with TOF in the present study.
Besides, individual changes linked to single lesions include the triple gain at 1q21.2 that was also implicated in TOF. Being a disorder involving more than one lesion, it is perhaps not surprising that TOF is extensively linked to genomic instability involving multiple loci. The question arises therefore as to whether several mechanisms and disease pathways may equally contribute to its manifestation. With respect to 1q21.2, in particular, it should be noted that, although CNVs in this region have been implicated in CHD, primarily TOF (Greenway et al., 2009; Liu et al., 2016), no specific gene appears to have been identified as the culprit yet. Potential candidates include the NBPF16, PPIAL4A- PPIAL4E, LOC645166, FCGR1C, and HIST2H2BF, among others.
The NBPF16 constitutes a member of the NBPF family consisting of dozens of duplicated genes primarily located in segmental CNGs on human chromosome 1, and the PPIAL4 gene family encodes a protein family that accelerates protein folding, both of which may influence developmental processes. The 2q13, which exhibits four different gains, harbors the NPHP1 and the LINC00116 genes. The NPHP1 encodes a protein with Src homology domain 3 (SH3) pattern that interacts with Crk-associated substrates, and appears to control cell division as well as cell/cell matrix adhesion signaling, possibly as part of a multifunctional complex localized in actin- and microtubule-based structures. CNVs in the NPHP1 have been implicated as a carrier state in CHD (Boone et al., 2013), whereas NAHR-mediated CNVs described for NPHP1 duplications are thought to be potential sources of diseases in general (Dittwald et al., 2013).
It is noteworthy that, in our study, the gains at the NPHP1 locus were associated with valvular lesions, aortic arch disease, as well as TOF, possibly pointing to a common pathway for the three lesions.
Unlike CNGs, the CNLs were found mostly singularly, except on chr 16p11.2 harboring three and 22q11.23 exhibiting two, respectively. The gains on 16p11.2 were linked to TP53TG3B and TP53TG3C (Zhu et al., 2018). A previous study has reported an association of 16p11.2 microduplication with TGA (Karunanithi et al., 2017). Interestingly, the losses on 22q11.23 occurred at the same genic loci as the gains. However, these losses were associated with septal and valvular diseases instead. This suggests that different genomic CNVs may be associated with several forms of septal defects occurring from different genomic mechanisms and gene interactions. Further genomic regions of interest include the chr 15q11.2, 19p13.3, and 20p12.3. The chr 15q11.2 harbors among others, the NIPA1, NIPA2, TUBGCP5, CYF1P1, and WHAMP3 (Lage et al., 2012).
NIPA1 encodes a magnesium transporter that may play a role in nervous system development and maintenance. Copy number alterations possibly encompassing the NIPA1 and CYF1P1 at this locus have previously been associated with sporadic CHD (Glessner et al., 2014; Soemedi et al., 2012). Put together, it is interesting to note that septal defects are linked to a couple of genes that are involved in structural functions. Furthermore, genomic sequence loss was almost equally linked to organism injury and abnormality as well as cardiovascular disease. It was also particularly enriched in dopamine degradation and signaling by Rho family GTPases as well as cytokine growth factor and transcription regulatory networks. These pathways are likely to explain some of the major mechanisms involved in disorders associated with CNLs, which need to be explored further.
Altogether, the genes discussed herein cover a part of the complex network likely to contribute to the pathogenesis of CHD in general. Particularly interesting was the manifestation of CHDs sharing multiple genomic risk sequences as indicated by several of the lesions being linked to the same genes, or the complex cases involving various forms of defects exhibiting deletions at multiple loci. Given this sharing of loci, the amount of convergence at such loci as well as the complex nature of CHD, it is tempting to speculate on different pathways ascribing to the manifestation of these lesions, dependent on their respective developmental pathways. The implications are that it may take a summation of all activities at a given locus to trigger a CHD phenotype. It also explains the complex nature of CHD phenotypic expression.
Therefore, the present findings demonstrate the impact of CNVs in CHD, the mechanisms of which require to be addressed deeper. In this regard, it is noteworthy that different platforms employed to test the impact of genomic changes in disease tend to yield varying results, depending on the target lesions and genotyping platforms. For example, with regard to CHD specifically, a GWAS by Cordell et al. (2013) described a susceptibility locus at chr4p16 for ASD, demonstrating different loci for risk variants compared with those affected by CNVs. A study by Lage et al. (2012) suggested some pattern of functional interactions between genomic variation and environmental exposure with modulating critical biological systems in cardiac developmental processes, adding weight to the likelihood of an environmental component to spontaneous CHD manifestation, for example.
Another investigation employing exome sequencing by Sifrim et al. (2016) described exclusive association of syndromic CHD with de novo variants in a number of familiar genes. These differences in the findings employing various platforms is an indication of the polygenic nature of CHD lesions being ascribable to a range of genomic sequences and genetic point mutations, thereby affecting systems which may or may not converge at various junctions in cardiac developmental processes.
Importantly, our study examined a whole pallet of CHDs, describing their links to genomic instability in various chromosomal regions affecting such important cardiovascular developmental systems and pathways. The literature purports that the larger the CNV and the greater the gene content, the more likely the variation will segregate with the phenotype and correlate with duplication pathogenicity, consistent with deleterious consequences from extra copies of many genes (Kearney et al., 2011).
Furthermore, it is thought that, because the same genes are deleted or duplicated, recurrent CNVs with common breakpoints can define genomic disorders characterized by particular clinical features (Watson et al., 2014). Besides, rare deletions or duplications have been linked to phenotypes of infants tested for CNVs (Cooper et al., 2011; Harley et al., 2008). Accordingly, our identification of several genes within long stretches of the affected genomic regions points to their potential relevance with respect to disease manifestation, and has important clinical implications for combating these disorders.
Collectively, therefore, although no direct clinical application can be derived from our data at this stage, the volume of genomic data yielded and the described systems being influenced by these genomic changes provide nonetheless a platform to be tapped as a basis for further exploitation in deciphering disease pathways leading to CHDs.
Conclusions
Our study reveals a high yield of CNVs in a broad spectrum of CHDs, primarily in the chromosomal regions of chr 17q21.31, 16p11.2, 8p11.21, and 22q11.23 in a large cohort of Saudi patients. Functionally, the observed changes were enriched primarily in genes and pathways involved in cardiovascular development and function, among others. The study reveals the essence of genomic instability in CHD, calling for further more in-depth evaluation of the potential relevance of CNVs in the management of these disorders.
Footnotes
Acknowledgments
This study was supported by the Royal Research Grant 2100020 (to Nduna Dzimiri) and Research grants (RAC#2110006 and 2180031 to Dr. Dilek Colak) through the King Faisal Specialist Hospital and Research Center (KFSHRC) to whom the authors would like to express their gratitude. They also would like to thank Dr. Namik Kaya for much helpful discussion and constructive remarks.
Author Disclosure Statement
The authors declare there are no conflicting financial interests.
Funding Information
This study was supported by the Royal Research Grant 2100020 (to Nduna Dzimiri) and Research grants (RAC#2110006 and 2180031 to Dr. Dilek Colak) through the King Faisal Specialist Hospital and Research Center (KFSHRC).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.