
Abstract
Coconut (Cocos nucifera L.), an important source of vegetable oil, nutraceuticals, functional foods, and housing materials, provides raw materials for a repertoire of industries engaged in the manufacture of cosmetics, soaps, detergents, paints, varnishes, and emulsifiers, among other products. The palm plays a vital role in maintaining and promoting the sustainability of farming systems of the fragile ecosystems of islands and coastal regions of the tropics. In this study, we present the genome of a dwarf coconut variety “Chowghat Green Dwarf” (CGD) from India, possessing enhanced resistance to root (wilt) disease. Utilizing short reads from the Illumina HiSeq 4000 platform and long reads from the Pacific Biosciences RSII platform, we have assembled the draft genome assembly of 1.93 Gb. The genome is distributed over 26,855 scaffolds, with ∼81.56% of the assembled genome present in scaffolds of lengths longer than 50 kb. About 77.29% of the genome was composed of transposable elements and repeats. Gene prediction yielded 51,953 genes, which upon stringent filtering, based on Annotation Edit Distance, resulted in 13,707 genes, which coded for 11,181 proteins. Among these, we gathered transcript level evidence for a total of 6828 predicted genes based on the RNA-Seq data from different coconut tissues, since they presented assembled transcripts within the genome annotation coordinates. A total of 112 nucleotide-binding and leucine-rich repeat loci, belonging to six classes, were detected. We have also undertaken the assembly and annotation of the CGD chloroplast and mitochondrial genomes. The availability of the dwarf coconut genome shall prove invaluable for deducing the origin of dwarf coconut cultivars, dissection of genes controlling plant habit and fruit color, and accelerated breeding for improved agronomic traits.
Introduction
Coconut (Cocos nucifera L., 2n = 32
Coconut oil is used extensively in soaps, cosmetics, paints, varnishes, detergent, surfactant, and emulsifier manufacturing industries (Siriphanich et al., 2011). The fibers (coir) obtained from husk by retting, which is one of the most rigid and most long-lasting natural ligno-cellulosic fibers known, is used in the making of ropes, mats, and carpets (Basu et al., 2015). Coconut shell-derived activated carbon find large scale use as a phenol absorbent (Freitas et al., 2019).
Many recent studies have attributed novel industrial uses to coconut products. Green coconut shells (Gonçalves et al., 2015) and green coconut fibers (da Costa Nogueira, 2019) have been reported to be alternative substrates for the production of bioethanol. Unsaturated polyester resins, a class of thermoset polymers developed from coconut oil, displayed greater thermal stability (Costa et al., 2016). da Fonseca et al. (2020) presented several advantages on the use of a biocatalyst, consisting of enzymes from tender coconut water immobilized onto hydrogel microcapsules (i.e., activated sodium alginate), on the bioacylation of quinine. Nanomaterials derived from coconut fibers (Juikar and Vigneshwaran, 2017) and fresh coconut inflorescence sap (Rajesh et al., 2020) could have potential textile and biomedical uses, respectively.
A member of the Arecaceae family, the coconut palm is monotypic, being the only species in the Cocos genus. Traditionally, coconuts have also been categorized into tall and dwarf varieties (Narayana and John, 1949), based on palm stature and breeding pattern. Tall populations occupy the major area under coconut worldwide with dwarfs accounting for only 5% of coconut populations. While the talls are largely outcrossing, the dwarf types are predominantly self-pollinating. Palms with intermediary growth habits are common and can include either natural or artificial hybrids (Arunachalam and Rajesh, 2008). Tall palms are preferred for their copra, coconut oil, and fiber, while dwarfs are sought out for their sweet tender nut water, resistance to phytoplasma disease, and ornamental value. Dwarfs are utilized in breeding programs with talls to create hybrids to exploit hybrid vigor (Nair et al., 2016).
The perennial nature of the palm, its long juvenile phase, inherent high levels of heterozygosity, the requirement of a large area of land for experimentation, and lack of techniques for mass vegetative propagation of palms with superior traits have been major impediments and create complications in breeding efforts (Nair et al., 2016). Availability of coconut genomic resources can revolutionize coconut breeding and aid in investigating and uncovering the complex pathways, which underlay phenotypic variations of traits of importance such as nut yield, copra content, oil yield, oil quality, dwarfism, fruit color, and disease resistance, among others.
The draft genome of a tall accession (Hainan Tall) has been made available (Xiao et al., 2017), with a scaffold length of 2.20 Gb (N50 = 418 kb). Also, while the analysis of the Chowghat Green Dwarf (CGD) genome was underway, the genome of a Catigan Green Dwarf, consisting of 2.1 Gb (N50 = 570.49 kb) sequence, has been published (Lantican et al., 2019).
The availability of multiple reference genomes of coconut would enable variant discovery and deciphering of the genetics of traits of agronomic importance. We have utilized massively parallel sequencing to assemble a draft genome sequence of an indigenous dwarf cultivar CGD. Less incidence of root (wilt) disease, which is a severe but debilitating disease affecting palms in India, have been recorded on CGD palms (Nair et al., 2004). CGD palms are self-pollinated; detailed studies carried out to assess the breeding behavior of these palms have revealed that there is complete overlapping of the male and female phases, in an inflorescence, implying facilitation of self-pollination. Evidence from microsatellite genotyping also points out the genetic uniformity of these palms (Thomas et al., 2015). Therefore, the CGD genome is expected to possess greater homozygosity, making it more suitable for sequencing and serve as a reference genome for coconut.
Materials and Methods
A schematic workflow of steps undertaken in this study is given in Figure 1.

The workflow highlighting the key steps used in this study for coconut genome assembly and annotation.
DNA isolation and flow cytometry
Genomic DNA of high quality was extracted from spindle leaves of a typical CGD palm (Accession IND029; IC296656), grown in the farm at Indian Council of Agricultural Research-Central Plantation Crops Research Institute, Kasaragod, Kerala, India, using DNeasy Plant Mini Kit (Qiagen, Germany). The genomic DNA was treated with RNaseA (Fermentas, USA) and proteinase K (Fermentas) to remove RNA and protein contamination, respectively, and the DNA was further precipitated with ethanol. The presence of high-molecular-weight DNA was visualized through 1% agarose gel electrophoresis stained with SYBR Safe (Invitrogen, USA). The DNA concentration was quantified using the Picogreen method in a Victor3 multilabel reader (Perkin Elmer, USA).
Before sequencing, the genome size of the CGD palm was determined using flow cytometry on a BD FACSCalibur flow cytometer, using Pisum sativum as an internal standard. BD CellQuest Pro software package was used to analyze the data. The genome size of CGD was estimated to be 2.59 Gb using flow cytometry.
Genome sequencing, assembly, and quality assessment
Deep sequencing of high-quality genomic DNA was performed using a hybrid sequencing strategy comprising short reads from Illumina and long reads from Pacific Biosciences (PacBio) platforms. Paired-end libraries of two insert sizes (300 and 600 bp) and mate-pair libraries of three insert sizes (3, 5, and 10 kb jumping distances) were prepared. The DNA sequencing was performed on an Illumina HiSeq 4000 sequencer with 2 × 101 bp sequence read length. The paired-end and mate-pair sequencing libraries were generated according to the standard protocols for the TruSeq Nano DNA LT Sample Prep Kit and the Nextera Mate Pair Sample Preparation Kit, respectively (http://illumina.com/).
Construction of the sequencing library for the PacBio sequencer was carried out utilizing the SMRTbell Template Prep Kit 1.0 (http://pacb.com/). The SMRTbell Template sequencing primer with DNA polymerase was applied to the single-molecule real-time (SMRT) Cell for the sequencing reaction. The P4 DNA polymerase with C2 chemistry (P4–C2; DNA/Polymerase Binding Kit P4, DNA Sequencing Reagent 3.0) was used in the sequencing reaction using the PacBio RS II sequencer to generate sequencing data with an average read length of 10 kb. Read correction and trimming of PacBio reads was undertaken using CANU assembler (Koren et al., 2017), and output CANU trimmed reads were used for further downstream analysis. The sequence data were analyzed using SMRT Analysis v.2.2.0 (PacBio).
The CGD genome was assembled, with the reads obtained from the two sequencing platforms, under two conditions. First, raw sequencing reads from the Illumina Platform was trimmed and filtered according to quality using NGS-Toolkit (Patel and Jain, 2012), by removing adaptor contaminations and low-quality reads with base N's or more than 70% of bases with a quality score <20. The draft de novo assembly of the whole genome sequencing (WGS) short reads from the Illumina platform was performed by SOAPdenovo2 (Luo et al., 2012).
The contigs generated from the primary assembly were subjected to scaffolding using SSPACE Standard (Boetzer et al., 2011) along with three Illumina mate-pair high quality (HQ) libraries. From the scaffolds generated using paired-end and mate-pair libraries, scaffolds of length <1 kb were removed to reduce the fragmented nature of the assembly. Filtered secondary scaffolds ≥1 kb were used to generate a secondary assembly by incorporation of PacBio long trimmed reads using SSPACE-LR scaffolder. Finally, the scaffolds generated from SSPACE-LR (secondary assembly) were subjected for rescaffolding using all the five Illumina paired-end (PE) and mate-pair (MP) libraries to generate the draft genome assembly of dwarf coconut cultivar.
Second, we also performed a hybrid genome assembly of short and long reads using Maryland Super Read Cabog Assembler (MaSuRCA) v3.3.1 (Zimin et al., 2013) with default parameters. Raw sequencing reads from the Illumina platform were trimmed and filtered using TrimGalore v 0.5.0 (Krueger, 2015) with a quality cutoff of 30, and other parameters kept default. After trimming off all the paired-end and mate-pair raw data, paired and unpaired reads from paired-end library size 600 bp (SRR6301636) with fragment standard deviation at 90 and only paired reads from paired-end library size 300 bp (SRR6301637) with fragment standard deviation at 45 were considered.
However, three jump libraries with a distance of 3 kb with fragment standard deviation at 450, 5 kb with fragment standard deviation at 750 and 10 kb with fragment standard deviation at 1500, were considered with only paired-end reads left out after the trimming process. Also, the set of 44 PacBio reads were merged for the assembly process.
Characterization of repeats and simple sequence repeats
The draft CGD genome assembly was screened for the identification of the repeat families. RepeatModeler v.1.0.8 (http://repeatmasker.org/RepeatModeler/), as well as “embryophyta” library, was made use of for the identification of de novo repetitive sequences in the coconut genome based on a self-BLAST search. RepeatMasker v.4.0.5 (http://repeatmasker.org/) was used to investigate known repetitive sequences utilizing a cross-match program with a Repbase-derived RepeatMasker library (v.20140131). De novo repetitive sequences were constructed using RepeatModeler. All the assembled contigs were screened for the presence of simple sequence repeats (SSRs) using MISA (Beier et al., 2017).
Quality assessment using BUSCO
BUSCO (Benchmarking universal single-copy orthologous genes) (Simão et al., 2015) was used for assessment of coverage of genes that are most probable to be present/absent in the scaffold as a measure of the completeness of the genome assembly. The evaluation was performed against the “embryophyta” gene sets.
Gene prediction
The repeat-masked assembly was used for annotation of the protein-coding genes using the automated gene prediction pipeline, SeqPing (Chan et al., 2017), which makes use of both Hidden Markov Models (HMM) and data from transcriptome analysis. Through this pipeline, genome and transcriptome sequences are first processed using three different pipelines viz., GlimmerHMM (Majoros et al., 2004), SNAP (Korf, 2004), and AUGUSTUS (Stanke and Morgenstern, 2005). The predictions from these three pipelines are then combined with transcriptomic evidence by MAKER (Cantarel et al., 2008; Campbell et al., 2014). SeqPing predicted genes were further filtered using quality_filter.pl with option-s (standard cutoff of annotation edit distance [AED] <1); later gene and protein sequences for the same were extracted using gff3_sp_extract_sequences.pl script from the GAAS annotation tools (https://github.com/NBISweden/GAAS).
Transcriptome sequences, derived from reference-based transcriptome assembly and de novo assembly, were also used as evidence to guide the annotation process. The procedure adopted is briefly given below:
Reference-based transcript assembly
Raw files of coconut transcriptome data were downloaded from the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) repository. These include transcriptome data generated by RNA-Seq of leaves of healthy (SRA accession number SRX436961; CT1) and root (wilt) diseased (SRX437650; CT2) palms (Rajesh et al., 2015, 2018) and embryogenic calli (SRX472157; CT3) (Rajesh et al., 2016). The raw files were mapped to the assembled genome using HiSAT2 (Kim et al., 2019).
Furthermore, transcript models were built from the three datasets using StringTie (Pertea et al., 2015). SeqPing predicted gene annotation file was used as a reference for creating the transcript models. The predicted proteins from SeqPing were used as a query to perform tblastn (Gertz et al., 2006) against transcript models. Proteins possessing a blast hit, with query coverage and percentage identity of ≥90%, were considered to have protein-coding evidence in the transcript models.
De novo transcript assembly
Proteome data obtained from SeqPing analysis were mapped against de novo assembled transcripts obtained from RNA-Seq of young endosperm tissue (SRX1719682; CT4) (Fan et al., 2013), leaves of healthy (SRA accession number SRX436961; CT5) and root (wilt) diseased (SRX437650; CT6) palms (Rajesh et al., 2015, 2018), leaves of Chowghat Orange Dwarf palm in response to infection by Phytophthora palmivora (SRR9140951; CT7) (Gangaraj and Rajesh, 2020), and embryogenic calli (SRX472157; CT8) (Rajesh et al., 2016) using tblastn (Gertz et al., 2006). Blast hits with 90% of query coverage and identity were considered to have evidence in the transcript dataset.
Identification of noncoding RNAs
In addition, tRNAscan-SE software (Schattner et al., 2005) was used with the default parameter setting to identify the tRNA genes. The sequences with an overall score of more than 50 were considered and a nonredundant set of the sequences was listed as tRNAs. The rRNA genes were identified using homology-based evidence by searching against SILVA database v 132 (Quast et al., 2012). Large subunit genes were downloaded from SILVA, and plant-specific sequences were extracted using an in-house python script.
The redundant sequences were clustered and removed using CD-HIT (Fu et al., 2012) at 90% identity. The nonredundant sequences were used to blast against the scaffolds. The hits with e-value <1.0E-5 were considered as rRNA. microRNA (miRNA) and small nuclear RNA (snRNA) were identified using the Infernal tool (Nawrocki and Eddy, 2013) with default parameters against the Rfam database (Kalvari et al., 2018) as a reference. The accepted nomenclature was followed for naming the miRNAs predicted from the CGD genome. The prefix “cnu” was added before the miRNA to denote
Functional annotation and pathway identification
For functional annotation, the predicted protein-coding genes were annotated using PANNZER2 (Törönen et al., 2018), a speedy functional annotation web server that delivers free text description predictions in addition to Gene Ontology (GO) annotations.
Identification of transcription factors, transcription regulators, and protein kinases in CGD genome
Identification and classification of transcription factors (TFs), transcription regulators, and protein kinases were carried out using iTAK (Zheng et al., 2016).
Identification of nucleotide-binding site-leucine-rich repeat gene family in the CGD genome
Prediction of nucleotide-binding and leucine-rich repeat (NLR) genes was undertaken using NLR-Annotator (Steuernagel et al., 2020) CGD genomic scaffolds were initially chopped into overlapping subsequences, which were scanned for sequence motifs, in all six frames, by using the MAST tool available under MEME suite (Bailey et al., 2009). In addition, the translated protein sequences were scanned specifically for NB-ARC domain (PF00931), TIR domain (PF01582) and RPW8 (PF05659) using PfamScan (http://ebi.ac.uk/Tools/pfa/pfamscan). A nonredundant catalog of putative NLRs (coiled-coiled-NBS-LRR [CNL], toll-interleukin-1 receptor-like-NBS [TN], coiled-coiled-NBS [CN], NBS-LRR [NL], and resistance to powdery mildew RPW8-NBS-LRR [RNL]) was then created by combining both the genome and proteome predictions.
Domain prediction of the individual NLRs was carried out by InterProScan (Jones et al., 2014) for further domain prediction. Orthologs, from oil palm and date palm, were identified by aligning the NLRs using MAFFT (Katoh and Standley, 2016). Phylogenetic analysis of the aligned data was performed using the maximum likelihood method (1000 bootstraps) in the MEGA Version 7.0.14 package (Kumar et al., 2016).
Assembly and annotation of chloroplast and mitochondrial genomes
The chloroplast genome of the CGD cultivar was assembled using NC_022417.1 as a reference. The CGD mitochondrial genome fragments were extracted from the scaffolds using BLASTN with KX028885 as reference. The fragments with ≥90% identity and alignment length of ≥300 bp were considered for the scaffolding using MeDuSa scaffolder V.1.6 (Bosi et al., 2015). The assembled chloroplast and mitochondrial genomes were annotated with GeSeq (Tillich et al., 2017) and the generation of its graphical map was carried out with the help of OGDRAW (Lohse et al., 2007).
Data availability
Raw nucleotide sequence data are available in the NCBI sequence read archive database (BioProject PRJNA413280) under the accession number SRS2696501.
Results
Sequencing and de novo assembly of the dwarf coconut genome
We have sequenced and assembled the genome of a dwarf coconut cultivar, CGD using two sequencing platforms. Sequencing on Illumina HiSeq 4000 platform generated 183.51 Gb of sequence data, representing ∼70.6 × genome coverage. Furthermore, 37.02 Gb of sequence data were generated on the PacBio platform representing ∼14.3 × genome coverage. Quality control of deep sequencing data obtained using different sequencing strategies resulted in more than 95% of the raw data possessing a Phred quality score of >30.
Assembly of the trimmed and high-quality reads, first undertaken using SOAPdenovo2, resulted in a primary assembly with a maximum contig length of 704 kb and N50 of 48 kb, in 571,161 scaffolds with a total genome coverage of 1.94 Gb. Length distribution of scaffolds showed that 481,794 (84%) of these scaffolds were <1 kb, totaling 161 Mb (∼8%) out of 1.94 Gb of assembly size. The primary reason for this high fragmentation could be the highly repetitive nature of the coconut genome as well as the use of only short-read sequencing platforms during the primary assembly.
All contigs/scaffolds below 1 kb were discarded from the primary assembly, leaving 89,469 scaffolds with a genome coverage of 1.78 Gb for further analysis, with an N50 of 55 kb and maximum scaffold length of 704 kb. The obtained scaffolds, along with PacBio corrected reads subjected to SSPACE-LR, resulted in the second assembly of 63,714 scaffolds with 1.84 Gb of genome coverage and N50 of 77 kb. By using Illumina mate-pair reads, along with the secondary assembly scaffolds, the assembly was further improved using SSPACE scaffolder and the final assembly of 59,328 scaffolds, with an N50 of 86 kb, was generated (Table 1). The final scaffold size of 1.84 Gb of the CGD draft genome assembly represents about 70% of the genome size of 2.59 Gb, as estimated by flow cytometry.
Comparison of Final Assembly Statistics of the Genome of Chowghat Green Dwarf Variety Using Two Assemblers
MaSuRCA, Maryland Super Read Cabog Assembler.
The final hybrid assembly, generated using MaSuRCA, resulted in 26,885 scaffolds with an N50 of 128.74 kb (Table 1). The final scaffold size of 1.93 Gb of the CGD draft genome assembly represents about 75% of the CGD genome size. The detailed metrics of the draft genome are provided in Table 2.
Metrics for the Draft Genome Assembly of Chowghat Green Dwarf Variety
The number of scaffolds, nucleotides, and N content are indicated for the different size range of assembled scaffolds.
Characterization of repeats and SSRs
A survey of repetitive elements in the assembly using RepeatModeler and RepeatMasker software revealed that the CGD genome has a relatively large proportion of (∼77.29%) repetitive DNA. Similar to other plant species, the long-terminal repeat (LTR) retrotransposons comprised the most abundant class of repetitive DNA (58.85%), with copia (36.80%) and gypsy (21.44%) being the most abundant elements. A complete list of transposable elements is presented in Supplementary Table S1.
A total of 281,666 SSR or microsatellite loci were also identified in the assembly, including di- (80,469), tri- (35,686), tetra- (19,549), penta- (8235), and hexanucleotide (1026) repeats (Table 3).
Statistics of Simple Sequence Repeats Identified in Chowghat Green Dwarf Genome
SSR, simple sequence repeat.
Quality assessment using BUSCO
Estimation of genome completeness using BUSCO resulted in the identification of a total of 1440 genes (84.6%) present in the draft genome, of which 1068 (74.2%) were complete matches with full-length proteins and 78 (5.4%) showed fragmented hits. Comparison of the BUSCO results with the other sequenced palms viz., tall coconut (C. nucifera; Hainan Tall cultivar), dwarf coconut (C. nucifera; Catigan Green Dwarf), African oil palm (Elaeis guineensis), American oil palm (E. oleifera), and date palm (Phoenix dactylifera), showed a significant similarity of the dwarf coconut genome with these palms indicating that the draft assembly was quite similar to other assemblies of palm genomes, implying the integrity and quality of the CGD draft genome (Table 4).
BUSCO Comparison of Sequenced Palm Genomes
BUSCO, benchmarking universal single-copy orthologous genes.
Gene prediction and annotation
The gene prediction yielded 51,953 genes, which upon filtering based on AED resulted in 13,707 genes that coded for 11,181 proteins (Supplementary Table S2).
Evidence from available transcriptome data
Reference-based transcript assembly
Assembly of transcript data using StringTie resulted in the generation of 62,322, 61,573, and 35,895 transcript models from CT1, CT2, and CT3, respectively. Blast search provided evidence for 6717 proteins in CT1, 6714 proteins in CT2, and 4926 proteins in CT3 (Fig. 2). A total of 7100 nonredundant proteins were found to have evidence in at least one of the three transcriptome datasets.

Transcript level evidence of the predicted proteins. The transcriptome data generated by RNA-Seq of leaves of healthy (SRA accession number SRX436961; CT1 and CT5) and root (wilt) diseased (SRX437650; CT2 and CT6) coconut palms (Rajesh et al., 2015, 2018), coconut embryogenic calli (SRX472157; CT3) (Rajesh et al., 2016), young endosperm tissue (SRX1719682; CT4) (Fan et al., 2013), and leaves in response to infection by Phytophthora palmivora (SRR9140951; CT7) were used.
De novo transcript assembly
De novo transcript assembly resulted in the identification of 3153 proteins in CT4, 3594 in CT5, 3445 in CT6, 6049 in CT7, and 3140 in CT8 data (Fig. 2). A total of 6828 proteins were found to have evidence in at least one of the five transcriptome datasets.
Identification of noncoding RNAs
Overall, we identified 1214 noncoding genes; 470, 463, 8, 118, and 155 were categorized as tRNAs, rRNAs, snRNAs, small nucleolar RNAs, and miRNAs, respectively. The details of noncoding RNAs are provided in Table 5 and Supplementary Tables S3A–C.
Annotation of Noncoding RNA in the Chowghat Green Dwarf Genome
miRNA, microRNA; snRNA, small nuclear RNA.
Functional annotation and pathway identification
Based on the functional annotation, 5980 GO terms were assigned to 5096 coconut proteins (Supplementary Table S4).
Identification of TFs, transcription regulators, and protein kinases
A total of 314 genes were identified as potential TFs and 104 genes as potential transcription regulators. Furthermore, 76 genes, belonging to the protein kinase family involved in signal transduction pathways, were identified. Genes coding for TFs could be categorized into 48 categories, where C2H2 (27 genes), NAC (23 genes), GRAS (22 genes), bZIP (19 genes), and C3H (18 genes) were the major classes found (Fig. 3). A total of 104 genes could be grouped as transcription regulators comprising of 17 categories, of which major categories were mTERF (21 genes), SWI/SNF-BAF60b (10 genes), IWS1 (8 genes), and SET (8 genes) (Fig. 3). Furthermore, a total of 76 protein kinase genes were identified and classified into 38 subclasses (Fig. 4).
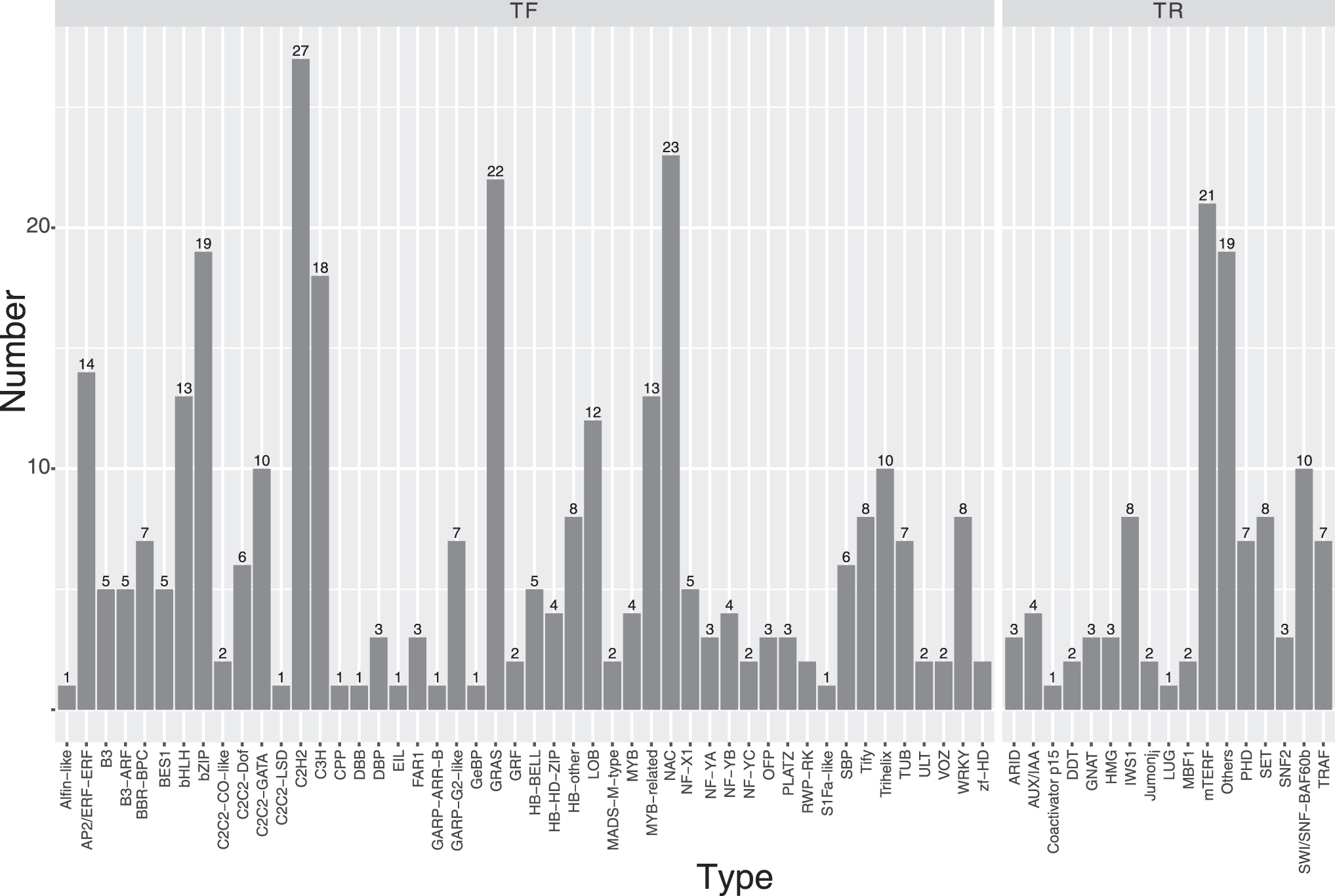
Categories of transcription factors and transcription regulators predicted in CGD genome. CGD, Chowghat Green Dwarf.

Categories of protein kinases predicted in CGD genome.
Identification of the NLR gene family in the CGD genome
A total of 112 NLR loci were identified in the CGD genome, which accounts for ∼0.8% of the 13,707 annotated genes. These NLRs could be classified into NBS–LRR (40 loci), CC–NBS–LRR (coiled-coiled NBS-LRR) (20 loci), NBS (29 loci), CC–NBS (20 loci), RPW8-NBS-LRR (RNL) (two loci), and TIR-NBS (TN) (one locus), based on the arrangement of the domains of the translated proteins. MEME analysis, undertaken on amino acid sequences of NBS domains of coconut NLRs, revealed highly conserved domains such as “GKTTLA” at P-loop, “GLPLA” at GLPLA, and “LLVLDDW” at Kinase 2 (Supplementary Table S5). Detailed phylogeny of NLR loci from the CGD genome is given in Figure 5. The phylogenetic results showed that the CC–NBS–LRR, TIR–NBS, RPW8–NBS–LRR, CC–NBS, and NBS–LRR formed the first clade, while NBS genes formed a separate, second clade.

Phylogeny of NLR loci in the CGD genome. Branch support values obtained are labeled on basal nodes. NLR, nucleotide-binding and leucine-rich repeat.
Assembly and annotation of chloroplast and mitochondrial genomes
The CGD chloroplast genome of 154,628 bp annotated to 129 genes, with 84 protein-coding genes, 38 tRNAs, and two copies each of the four rRNAs (Fig. 6; Table 6). With a 37.45% GC content, the CGD chloroplast genome possessed a typical plant chloroplast overlay with a standard quadripartite structure consisting of two inverted repeats (IRa and IRb), each spanning 26,525 bp region and the large single copy and small single copy regions comprising of 84,231 bp and 17,400 bp, respe (Fig. 6). Mitochondrial assembly of 744,799 bp, with a GC content of 41.86%, resulted in 123 genes, and the detailed information is provided in Figure 7 and Table 7.

Map of the CGD chloroplast genome with 129 annotated genes and showing well-defined boundaries of IR, SSC, and LSC regions. Genes on the outer side of the circle are transcribed clockwise, while those on the inside are transcribed counter-clockwise. The inner circle with a dark gray plot indicates the GC content of the chloroplast genome. Genes containing introns are marked with *. LSC, large single copy; SSC, small single copy; IR, inverted repeat.
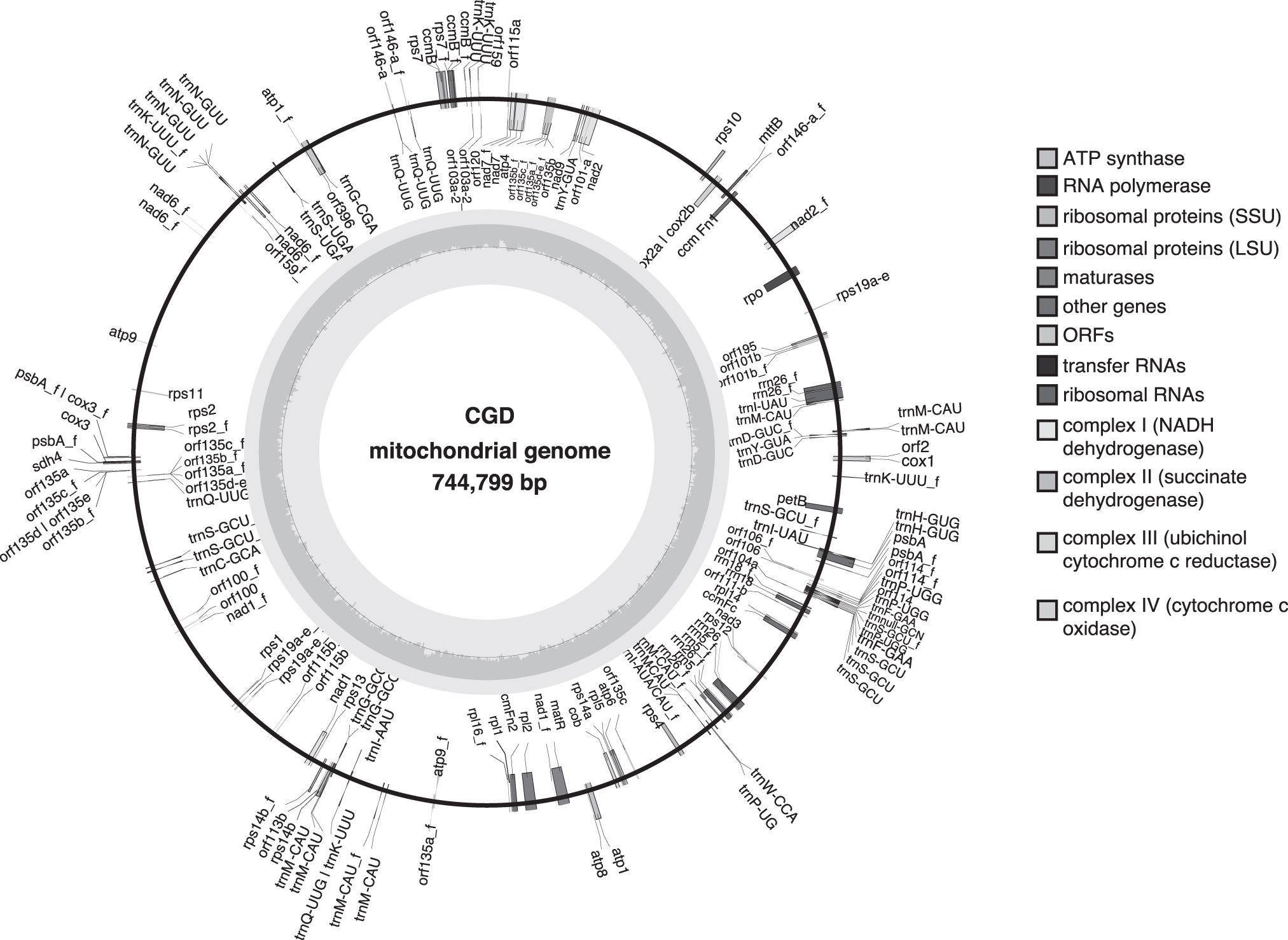
Circular map of the CGD mitochondrial genome. Features on transcriptionally clockwise and counter-clockwise strands are drawn on the inside and outside of the outer circle, respectively. The inner circle indicates the GC content as a dark gray plot.
Gene Annotation of the Chowghat Green Dwarf Chloroplast Genome
The numbers in brackets indicate the number of copies of the particular gene. ORFs, open reading frames.
Gene Annotation of the Chowghat Green Dwarf Mitochondrial Genome
The numbers in brackets indicate the number of copies of the particular gene. Fragmented genes are named with suffix “_f”.
SSU, small subunit; LSU, large subunit.
Discussion
Plant omics research offers veritable prospects toward applications in preventive medicine and health-related applications. Despite its long-standing importance, the coconut industry today is beleaguered with many problems compounded by the decline in production and productivity resulting in low income from coconut farming. Research on coconut as an oilseed crop is yet to reach the majority of producers who are small and marginal holders. Bridging this gap necessitates a novel approach that brings together researchers and other stakeholders. The complexities of coconut breeding and the limited choice of genomics tools available to date have hindered the ability of coconut breeders to exploit novel methods for crop improvement.
The genome assembly of palms such as coconut, possessing a large genome, coupled with excessive repetitive DNA, is challenging. Therefore, Illumina sequence data, using a series of paired-end and mate-pair libraries, and PacBio data were utilized for a hybrid genome assembly. The initial assembly, undertaken using SOAPdenovo2 and SSPACE resulted in a final scaffold size of 1.84 Gb, representing 70% of the genome size. An improved draft genome assembly was obtained using MaSuRCA, with a total size of 1.93 Gb, presents 75% of the dwarf coconut genome with N50 of scaffolds around 128.74 kb. The MaSuRCA, which is based on efficient de Bruijn graph methods and has been reported to be more efficient than SOAPdevovo2, relies on the generation of a lesser number of lengthier “super reads” from a large number of raw sequencing reads and can undertake hybrid assemblies of sequencing data generated from multiple platforms (Zimin et al., 2013).
Recently, MaSuRCA has been utilized for hybrid assembly (short Illumina reads and long PacBio reads) of the complex bread wheat genome (Zimin et al., 2017). The guanine-cytosine content of the CGD genome (37.59%) is quite similar to other palm genomes, including coconut (Catigan Green Dwarf [37.64%]), oil palm (36.8%) (Jin et al., 2016), and date palm (38.5%) (Al-Dous et al., 2011).
A total of 1.49 Gb of repeat elements were identified in a 1.93 Gb genome assembly, indicating that 77.29% of the assembled genome is repetitive, which is expected given the proportionality of repeat content with genome size. Since the repetitive portion of the genome always comprises underrepresented portions in the genome assembly (Varshney et al., 2017), it could be assumed that most of the unassembled DNA in coconut would most likely be repetitive too, making it challenging to assemble utilizing our sequence dataset and current assembly scheme. According to Liu and Bennetzen (2008) and Veeckman et al. (2016), during the process of assembly, multiple repeats most frequently collapse into a single repeat, in addition to the fact that “repeat masking” is executed before certain assembly procedures.
The proportion of repetitive elements in dwarf coconut is similar to the proportion of repetitive DNA reported in Hainan Tall coconut (>72.75%; Xiao et al., 2017) and Catigan Green Dwarf (∼78.33%; Lantican et al., 2019), but much higher than that reported for other palm genomes viz., 42.18% in dura oil palm (E. guineensis; Jin et al., 2016), 57% in pisifera oil palm (E. guineensis; Singh et al., 2013), 42.31–50.96% in American oil palm (E. oleifera; Singh et al., 2013), and 33.67% in date palm (P. dactylifera) (Filho et al., 2017).
A comparative analysis of the repertoire of repetitive elements in the dwarf coconut genome with other palm genomes showed that LTR retrotransposons is the most abundant class of repetitive DNA (58.85%). Among LTRs, the most abundant elements were copia (36.80%) and gypsy (21.44%), which is typical of monocot genomes (Du et al., 2010). We assume that slow, steady, and long-term accumulation of retrotransposons might have contributed to genome instability in coconut. Xia et al. (2017) had attributed amplification of LTR retrotransposons families to the decrease in efficiency of DNA removal mechanisms (both unequal homologous/illegitimate recombinations) in the tea tree genome, in comparison to Arabidopsis thaliana (Devos et al., 2002) and Oryza sativa (Devos et al., 2002; Ma and Bennetzen, 2004).
In contrast to the high proportion of unknown elements in the genome of Elaeis spp. (Filho et al., 2017), dwarf coconut genome contained only 11.65% of unknown elements. Many pieces of evidence have been put forth recently on the undeniable role of transposons in structuring genomes and also in genome evolution and epigenetics; hence, characterization of transposable elements assumes importance (Daccord et al., 2017).
BUSCO analysis has indicated the completeness of predicted genes in coconut. SeqPing gene prediction yielded 62,498 genes, which upon filtering based on AED resulted in 13,707 genes that coded for 11,181 proteins. More than 90% of quality trimmed reads from previous RNA-seq analysis (Fan et al., 2013; Gangaraj and Rajesh, 2020; Rajesh et al., 2015, 2016, 2018) could be successfully mapped back to our genome assembly. This could be construed as additional evidence of the completeness of dwarf coconut (CGD) genome assembly as the results are comparable with the expected mapping rates for RNA-seq data from other assembled genomes, both human and plant genomes, with 70–90% RNA-seq read mapping degree (Conesa et al., 2016; Zhuang and Tripp, 2017).
In addition to protein-coding genes, we have identified genes for noncoding RNAs in the draft dwarf coconut genome—a total of 470 genes for tRNAs and 463 genes for rRNAs were detected. The number of tRNA genes is similar to the 445 tRNA genes reported for date palm, but lesser than 636 predicted for oil palm (Jin et al., 2016).
TFs play a major role in stress signaling by regulating the expression of stress-inducible genes and therefore represent a vital component of plant stress signaling networks (Muthamilarasan et al., 2015). Plant TFs have been categorized into 58 families (Jin et al., 2014) and around 7% of plant genome has been reported to encode TFs (Udvardi et al., 2007). We have identified 1608 potential TFs in the CGD genome, with a majority of them belonging to the APETALA2/ethylene response factor (AP2/ERF) superfamily, which is one of the largest families of plant-specific TFs (Song et al., 2013).
The AP2/ERF TF family has been implicated in the regulation of diverse growth and developmental processes in plants, including responses to various stresses (Xu et al., 2011). Even though the genome-wide analysis of AP2/ERF TFs has been undertaken in many dicots, such studies are limited with respect to monocots (Wuddineh et al., 2015). The TF identified in the CGD genome could be potential candidates for delineating their roles in stress signaling and would aid the development of climate-smart varieties.
Diseases, especially the root (wilt) disease and lethal yellowing disease caused by phytoplasma, are a major constraint to coconut production and productivity. Given the biology of the crop, accurate diagnosis and identification of disease-resistant/tolerant palms is a herculean task. An analysis of NBS–LRR genes in the CGD genome revealed a total of 112 loci, including 40 NBS–LRR loci, 20 CC–NBS–LRR loci, 29 NBS loci, 20 CC–NBS loci, 2 RPW8-NBS-LRR loci, and a single TIR-NBS locus. The number of NLRs, even though comparable to other palms (Jin et al., 2016), are much fewer in comparison to other plants such as rice (Goff et al., 2002), sorghum (Paterson et al., 2009), and maize (Schnable et al., 2014).
We assume that similar to oil palm (Jin et al., 2016), NLRs in the CGD genome display more diversity, which along with their rapid evolution, would have enabled enhanced protection from challenges of diverse phytopathogens despite their relatively low numbers. We could not detect loci encoding for TIR-NBS-LRRs (TNLs), an observation consistent with the hypothesis that complete loss of TNL loci occurred early during the divergence of the monocot lineage (Shao et al., 2019; Tarr and Alexander, 2009).
However, we could detect a single gene encoding TIR-NBS (TN), corroborating the report of TN loci in the genome of Catigan Green Dwarf of coconut (Lantican et al., 2019). CGD cultivar has been identified to be tolerant to root (wilt) disease of coconut. The candidates NLRs identified offer possibilities for allele mining and design and validation of DNA markers for disease resistance in coconut breeding programs. Further functional characterization of these genes would permit deciphering the molecular mechanisms of coconut disease-resistant response to specific pathogens infecting coconut palms.
The organellar genomes (chloroplast and mitochondria) can offer plenty of molecular information to assist comparative evolutionary studies. It is easier to obtain mitochondrial or plastid genome sequences from total DNA by utilizing high-throughput sequencing data, in contrast to conventional approaches, wherein mitochondrial and plastid genome sequences are generated using separate procedures (Hao et al., 2014). Using reference-based assembly techniques, we have fished out chloroplast and mitochondrial reads from assembled CGD scaffolds and assembled them successfully into circular contigs. The chloroplast genome of coconut has been reported to be the smallest accounted so far in palms (Huang et al., 2013). The chloroplast genome of CGD cultivar, a typical quadripartite molecule, was exactly similar to the one reported earlier in dwarf coconut (Huang et al., 2013).
The CGD mitochondrial genome was also similar to the mitochondrial genome of Oman local Tall cultivar reported by Aljohi et al. (2016), with minor rearrangements. Organellar genomes, in general, possess slow nucleotide substitution rates compared to nuclear genomes and therefore bestow a suitable window of resolution to undertake in-depth studies of palm phylogenies at deeper evolutionary time scale levels. A comparison of the organellar genomes of tall and dwarf coconut cultivars could provide insights into the origin of dwarf coconuts and modifications that might have occurred during the process of domestication of coconut.
Conclusions
The availability of the draft genome of dwarf coconut cultivar would provide researchers with an invaluable resource that would facilitate a better comprehension of underlying trait variations and hasten coconut genetic improvement. It also offers a foundation for expeditious use of potent technologies such as genomic selection, for accelerating genetic gains in coconut. Considering over 26,885 scaffolds for the draft CGD genome, the impending task would be to fill in the gaps and connect scaffolds to generate a well-assembled genome.
The genome assembly reported here provides a draft “genomic blueprint” of a disease-resistant dwarf cultivar for coconut researchers, which should catalyze new applications toward coconut breeding programs and planetary health.
Footnotes
Authors' Contributions
P.C., R.K.M., T.S.K.P., A.K., V.N., N.K.S. and K.G. designed and executed the project; R.K.M. prepared the samples and extracted DNA for sequencing; S.K.B., S.K., R.B.S., G.B., A.J.D., R.K.M., and K.P.G. generated, curated, and analyzed the assemblies and annotation data; C.N.K., B.N., A.K.M., and V.M. carried out gene annotations and conducted the manual inspection of gene models; S.K.B., S.K., R.B.S., G.B., and A.J.D. analyzed the transcriptome data; S.K.B., S.K., V.M., K.P.G., and R.K.M. drew figures; S.K.B., S.K., V.M., B.N., C.N.K., and R.K.M. prepared tables and supplementary files; R.K.M,, S.K.B., S.K., B.N., and V.M. wrote the article with significant intellectual contributions from all authors. P.C., T.S.K.P., V.N., A.K., N.K.S., and K.G. revised the article. All authors have read and approved the final article.
Acknowledgments
S.K.B. is the recipient of the DBT-BINC Junior Research Fellow from the Department of Biotechnology, Government of India. S.K. is a recipient of a Senior Research Fellowship from the Indian Council of Medical Research (ICMR), Government of India. V.M. is a recipient of the Women Scientist-A award from the Department of Science and Technology, Government of India.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was supported by funding from the Indian Council of Agricultural Research (Indian Council of Agricultural Research-Central Plantation Crops Research Institute Project No. 1000761030).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.