
Abstract
About a tenth of all cancers are caused by viruses or associated with viral infection. Recent global events including the coronavirus disease-2019 (COVID-19) pandemic means that human encounter with viruses is increased. Cancer development in individuals with viral infection can take many years after infection, demonstrating that the involvement of viruses in cancer development is a long and complex process. This complexity emanates from individual genetic heterogeneity and the many steps involved in cancer development owing to viruses. The process of tumorigenesis is driven by the complex interaction between several viral factors and host factors leading to the creation of a tumor microenvironment (TME) that is ideal and promotes tumor formation. Viruses associated with human cancers ensure their survival and proliferation through activation of several cellular processes including inflammation, migration, and invasion, resistance to apoptosis and growth suppressors. In addition, most human oncoviruses evade immune detection and can activate signaling cascades including the PI3K-Akt-mTOR, Notch and Wnt pathways associated with enhanced proliferation and angiogenesis. This expert review examines and synthesizes the multiple biological factors related to oncoviruses, and the signaling cascades activated by these viruses contributing to viral oncogenesis. In particular, I examine and review the Epstein–Barr virus, human papillomaviruses, and Kaposi's sarcoma herpes virus in a context of cancer pathogenesis. I conclude with a future outlook on therapeutic targeting of the viruses and their associated oncogenic pathways within the TME. These anticancer strategies can be in the form of, but not limited to, antibodies and inhibitors.
Introduction
About a tenth of all cancers are caused by viruses and ∼80% of them occur in low income countries (Akram et al., 2017; De Martel et al., 2012; Schiller and Lowy, 2014). There are many challenges associated with diagnosis and treatment of cancers caused by viruses and these include the lack of good models to study the cancers as well as complex virus–host interactions during oncogenesis (Zur Hausen, 2009c). Although cancer-causing viruses are necessary for cancer development, they are not adequate for carcinogenesis (Bouvard et al., 2009; Zur Hausen, 2009c). Viruses appear to provide the initial “spark” needed for cancer formation with several other steps that do not involve viruses. Additional processes needed for cancer development after the initial virus infection may include carcinogens, inflammation, and immunosuppression (Fig. 1) (Bouvard et al., 2009; Zur Hausen, 2009c).

The involvement of viruses in cellular process.
This expert review examines and synthesizes the multiple biological factors related to oncoviruses, and the signaling cascades activated by these viruses contributing to viral oncogenesis. In particular, I examine and review the Epstein–Barr virus (EBV), human papillomaviruses (HPV), and Kaposi's sarcoma herpes virus (KSHV) in a context of cancer pathogenesis.
Role of Viruses in Carcinogenesis
Although viruses have been associated with several cancers, they are only a requirement in only a few cancers such as HPV in cervical cancer and KSHV in Kaposi's sarcoma (Moore and Chang, 2010; Zur Hausen, 2009b). Thus, although the prevalence of viruses might be high in individuals, cancers resulting from these viruses are always lower. In addition, cancers associated with viruses develop after a prolonged period of infection, with estimates given as 10–40 years after initial infection (Zur Hausen, 2009b).
However, as reported by Zur Haussen (2009b) lymphoproliferative disease associated with EBV infection can occur immediately after infection. The immune reaction to the presence of viruses can cause increased inflammation, thereby promoting cancer formation. Infection with a virus results in the virus hijacking the host's signaling system and promoting cellular division, survival, and metabolic reprogramming. In addition, the virus also prevents apoptosis from taking place to allow virus production. All these strategies are necessary for proper viral production and survival.
Several studies have shown that human oncoviruses or cancer-causing viruses hijack the cell signaling cascades to advance viral replication and survival. For example, the EBV-associated with several lymphomas and carcinomas, has been shown to imitate B cell proliferative signaling (Mancao and Hammerschmidt, 2007; Pattle and Farrell, 2006). This allows the EBV to replicate and be able to remain within cells. By inducing B cell proliferation so as to maintain EBV episomes replication, EBV can induce cancer formation (Burdin et al., 1993; Calender et al., 1987; Kempkes et al., 1995). Uninfected B cells can be removed by cytotoxic T lymphocytes, allowing infected B cells to survive and persist.
Most viruses utilize similar pathways to promote the process of tumorigenesis, with the p53 and retinoblastoma tumor suppressor pathways abrogated (Levine, 2009; Shackelford and Pagano, 2004). Included in the pathways targeted by viruses during tumorigenesis are the survival pathways PI3K-Akt, JAK-STAT, and the Wnt-B catenin (Bergonzini et al., 2010; Buchkovich et al., 2008; Fujimuro et al., 2003). In addition, different viruses are able to avoid detection or overcome recognition by the DNA damage response machinery of their host (McFadden and Luftig, 2013). The host cell is able to stop cellular division to finish DNA repair or until viral DNA has been cleared. Viruses are able to inactivate the host DNA repair mechanisms including apoptosis (Bartek et al., 2007; McFadden and Luftig, 2013).
With advances in cancer treatment, it is a requirement that viral DNA is checked during diagnosis and that any detected viruses and their associated pathways be targeted in tumors (Goldie et al., 2004; Schiller and Lowy, 2014). Vaccines for several viruses are also available for the prevention of infections, but more therapeutic options are clearly needed.
Molecular Mechanisms of EBV Oncogenesis
EBV displays patterns of latency associated with distinct diseases. During latency I, associated with Burkitt's lymphoma, EBV episomes are attached to the chromosomes of the host cell (Fig. 2). This enables the episomes to be retained during cell division. The expression of EBV nuclear antigen 1 (EBNA-1) is necessary to prevent cell death and thus enable survival of the EBV (Kirchmaier and Sugden, 1997). During latency II, associated with Hodgkin's disease and carcinomas, EBV oncogenes latent membrane protein (LMP)1 and LMP2A are expressed and these copy proliferative cues from B cells (Fig. 2) (Merchant et al., 2000; Mosialos et al., 1995). LMP1 causes the activation of signaling cascades including the nuclear factor-κB (NF-κβ), which is needed for lymphomagenesis (Mosialos et al., 1995). NF-κβ signaling activation results in cancer cell survival through upregulation of survival proteins including B cell lymphoma 2 (BCL-2) (Rowe et al., 1994).
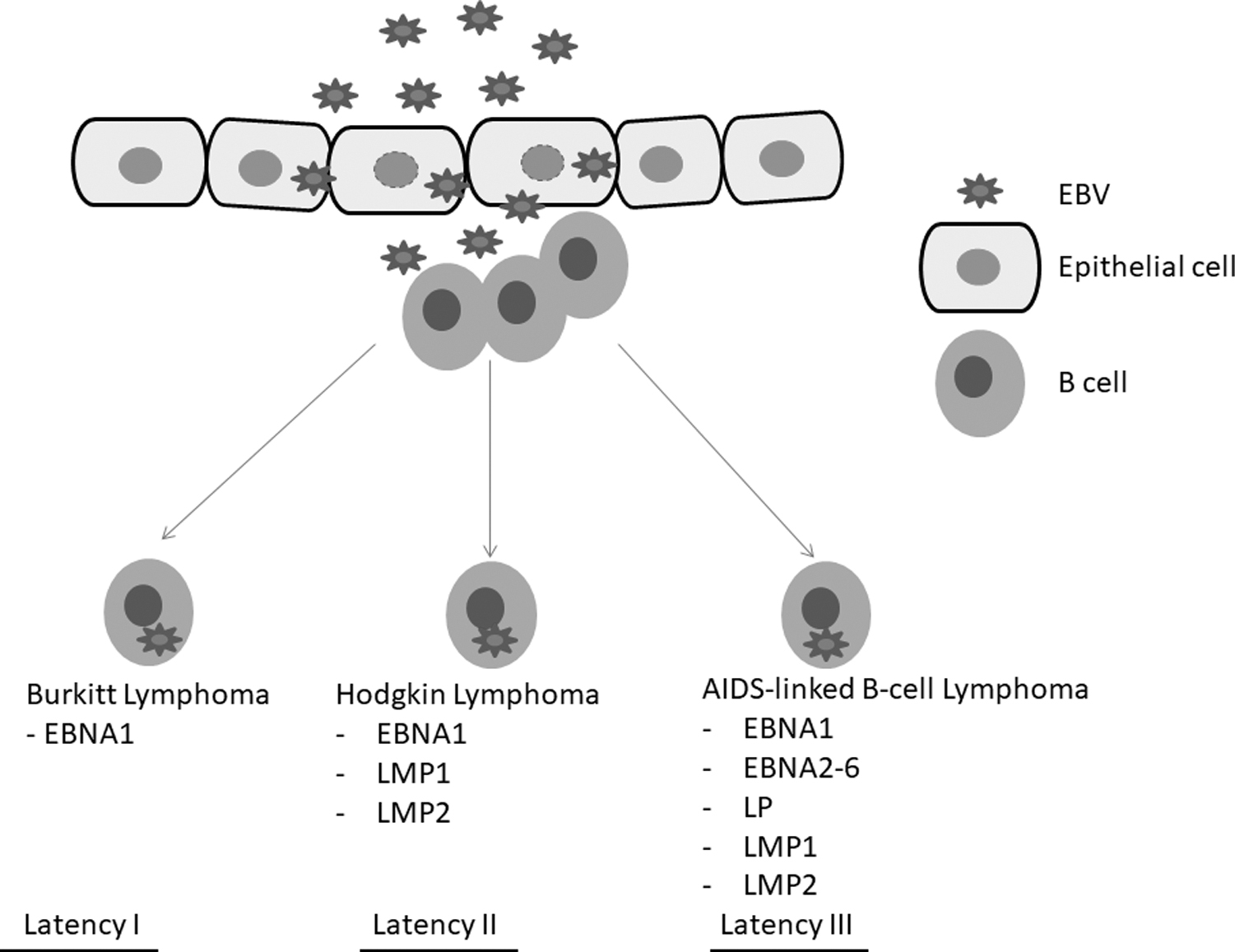
Different lymphoproliferative disorders display distinct patterns of Epstein–Barr virus latent gene expression. Figure adapted from Hui et al. (2019).
Several other genes associated with B cell malignancy and cellular invasive behavior are also upregulated and these include CD40 and interleukin (IL)-6 (Chang et al., 2005). LMP1 is also known to activate telomerase and thus promote genetic instability (Mei et al., 2006). On the contrary, LMP2A is able to imitate B cell Ig receptor signaling (Küppers, 2003). In the process, LMP2A enhances B cell differentiation and survival through the activation of signaling pathways including the PI3K-Akt pathway (Incrocci et al., 2017; Morrison et al., 2003).
EBV latency III is associated with acquired immunodeficiency syndrome (AIDS)-linked non-Hodgkin's lymphomas and has been shown to express several nuclear proteins linked to oncogenesis. These nuclear proteins include EBNA2, EBNA3, and EBNALP (Cooper et al., 2003; Longnecker et al., 1993). EBNA2 protein activates the expression of genes associated with Notch signaling leading to the dysregulation of the pathway (Höfelmayr et al., 2001). Several lymphoid malignancies have been associated with dysregulated Notch signaling (Mirandola et al., 2011; Vázquez-Ulloa et al., 2018). EBNA2 has also been shown to downregulate cell cycle proteins such as p21 and upregulating cyclin Ds linked to cell proliferation (Hui et al., 2019; Tsurumi et al., 2005). The retinoblastoma tumor suppressor is degraded through the engagement of EBNA3C to the ubiquitin ligase complex (Knight et al., 2005).
Several studies have shown that EBV-infected cells also express lytic cycle genes especially those encoding IL-10 and BALF1 (Kutok and Wang, 2006; Thompson and Kurzrock, 2004). Arvey et al. (2012) demonstrated that EBV lytic genes can be coexpressed with several cancer-linked cascades in EBV-infected cells. Another study demonstrated that autophagy, which is necessary to remove pathogens and maintain cellular homeostasis, is increased during the early stages of EBV lytic activation (De Leo et al., 2015). These studies and others demonstrate the possible involvement of lytic genes in oncogenesis. In addition, EBV is known to encode micro-RNAs. Two clusters of micro-RNAs are known to be located close to several EBV genes and are expressed in many B cells (Cai et al., 2006b; Klinke et al., 2014). These micro-RNAs can regulate many signaling cascades involved in cellular proliferative and apoptotic pathways (Cristino et al., 2019; Lin et al., 2015).
EBV-Associated Lymphomas
Several studies have shown that EBV latency II and III are oncogenic in several cells such as B and epithelial cells (Münz, 2019; Tsao et al., 2015; Yin et al., 2019; Young and Murray, 2003). This elicits an immunological response that is able to eliminate B cells infected with EBV in persons with competent immune systems (Alosaimi et al., 2019; Latour and Fischer, 2019). Such persons are therefore likely to display latency I and II in lymphomas; on the contrary, immunocompromised individuals will display latency III in lymphomas (Shannon-Lowe and Rickinson, 2019; Vockerodt et al., 2015). Cancer development mediated by EBV requires several cofactors such as chromosomal translocation and external factors such as infection with Plasmodium falciparum (Rochford et al., 2005; Van Tong et al., 2017; Vockerodt et al., 2015).
It is known that infection with P. falciparum results in polyclonal B cell proliferation, which is linked to chromosomal translocations in B cells, infected with EBV (Thorley-Lawson et al., 2016; Whittle et al., 1984). Chromosomal translocations in B cells may cause MYC expression dysregulation causing cell proliferation and metabolic reprogramming (Rochford et al., 2005; Tarrado-Castellarnau et al., 2016).
Immunosuppression is known to contribute toward the development of malignancies with latency III features and occur in human immunodeficiency virus (HIV)-associated non-Hodgkin's lymphomas. Individuals undergoing transplant and therefore with immunosuppression can have EBV-infected immune cells proliferating at a higher rate than normal and these cells can also acquire mutations (Bollard et al., 2012; Murray and Ibrahim, 2018). HIV-associated non-Hodgkin's lymphomas is complex but the occurrence of these disease is decreasing in individuals under antiretroviral therapy (ART) treatment (Grulich et al., 2001).
Other important factors for the development of latency III-associated malignancies include cytokine dysregulation and several HIV proteins (Casper, 2011). Nasopharyngeal carcinoma is an epithelial cancer associated with latency II expression patterns, with LMP1 being one EBV protein linked to tumor formation (Raab-Traub, 2002). Other factors contributing to the formation of nasopharyngeal carcinomas are linked to the environment and therefore not similar in all regions.
Human Papillomaviruses
Estimates indicate that ∼5% of human cancers are linked to HPVs. These viruses are not enveloped and are a group of viruses with DNA genomes that are double stranded and circular (De Villiers, 1989; Lambert, 1991). The HPV genome is composed of genes within the “early coded region” and others in the “late coded region” as well as a noncoding region (Handisurya et al., 2009). HPV has been described as the most common sexually transmitted virus causing several diseases. Approximately 200 genotypes of HPVs have been identified and only a small portion is considered “high risk” that is capable of infecting epithelial cells leading to tumor formation. HPV types can be classified into five genera specifically μ, β, α, γ, and ν, with the β and γ genera known to infect mostly the skin, whereas the α genera infects the mucosa (Brianti et al., 2017; Doorbar et al., 2012).
Reports indicate that almost all cervical cancers contain the α HPV DNA (Wang et al., 2020; Wu et al., 2018). About half of all infectious cancers in women are caused by HPV, whereas ∼5% of infectious cancers in men are caused by HPV (Zur Hausen, 2009a). In most cases, the human body is able to clear the HPV infection within 2 years after infection.
The most common “high-risk” HPVs are HPV16 and HPV18. In addition, these “high-risk” HPVs can cause anal, vulva, and oral carcinomas (Münger et al., 2004; Paz et al., 1997; Walboomers and Meijer, 1997). Low-risk HPVs are associated with the formation of benign warts (Ault, 2006; Chelimo et al., 2013). The most common low-risk HPVs are HPV6 and HPV11. Infected epithelial cells maintain a low copy number of viral genomes, with viral gene multiplication and expression limited to certain layers of the epithelium (Bedell et al., 1991; Wang et al., 2009). Through mechanisms including making differentiated keratinocytes be able to synthesize DNA, HPVs are able to maintain viral genome amplification in differentiated cells, and this may contribute to cancer formation (Mwapagha et al., 2017).
Reports indicate that most cervical carcinomas have HPV sequences integrated within the human genome and can lead to upregulation of genes such as MYC (Couturier et al., 1991; Dürst et al., 1987; Popescu and DiPaolo, 1989). In most cases, the integration of the HPV sequences results in dysregulated expression of the E6 and E7 proteins (Akagi et al., 2014; McBride and Warburton, 2017; Warburton et al., 2018). It is important to note that dysregulation of E6 and E7 protein expression can also result from epigenetic mechanisms (Hyland et al., 2011; Minarovits et al., 2016).
HPV carcinogenesis
HPV genes that produce oncoproteins include E5, E6, and E7. Cervical carcinomas that are HPV positive mostly express E6 and E7 proteins, referred to as high risk, that is known to induce senescence in several targeted signaling pathways (DeFilippis et al., 2003; Gupta et al., 2018; Pal and Kundu, 2020). HPV E5, E6, and E7 are all involved in the avoidance of immune detection and destruction. It is known that E5 proteins are able to reduce major histocompatibility complex class I expression and thus prevent recognition by CD8 T cells (Ashrafi et al., 2006; Campo et al., 2010). Both E6 and E7 are able to infect epithelial cells over a long time and in doing so avoids immune detection (O'Brien and Campo, 2002; Stanley, 2006).
Several signaling pathways involved in viral detection and destruction such as interferon (IFN) can be inhibited by both high-risk HPV E6 and E7 (Pol and Klingelhutz, 2013; Roman and Munger, 2013). Both E6 and E7 have been reported to be involved in tumorigenic-associated process such as angiogenesis and epithelial to mesenchymal transition (EMT). Duffy et al. (2003) demonstrated the involvement of E6 in increase in mesenchymal- and inflammation-associated genes. Evidence for the involvement of E7 in EMT was shown by Hellner et al. (2009).
Hallmarks of EMT included increased expression of vimentin and fibronectin and decreased expression of E-cadherin (Hellner et al., 2009). The expression of both E6 and E7 was associated with angiogenesis in several studies (Hoppe-Seyler et al., 2018; Li et al., 2011; Yeo-Teh et al., 2018). Expression of both E6 and E7 is also known to alter cellular signaling and metabolism, favoring cellular metabolic activity needed for viral survival (Spangle and Munger, 2013). E7 has been shown to direct the degradation of retinoblastoma tumor suppressor, allowing even differentiated cells to continue proliferating (Gonzalez et al., 2001; Liu et al., 2006). Although E7 expression can induce senescence, the removal of retinoblastoma tumor suppressor allows cells to continue proliferating by avoiding growth suppressors.
HPV E6 also induces the degradation of p53 through the action of ubiquitin ligase (Martinez-Zapien et al., 2016; Thomas et al., 1999). HPV E6 has also been shown to activate telomerase activity, allowing cells to proliferate indefinitely (Mwapagha et al., 2017). Reports indicate that HPV 16 E6 and E7 can preserve human epithelial cells in a proliferative state. Whereas immortalization itself does not mean the epithelial cells are oncogenic, continuous dividing can result in result in genomic I stability and mutations, and therefore produce tumorigenic cells (Hellner et al., 2009).
Not all cells infected by HPV will undergo malignant transformation. Several pieces of data show that viral transformation-associated proteins are strictly regulated (Cripe et al., 1987; Zheng and Baker, 2006). The chances of oncogenic transformation of cells are increased through uncontrolled expression of HPV proteins including E6 and E7 as well as viral genome epigenetic changes (Kgatle et al., 2017; Mwapagha et al., 2017).
Hepatitis B Virus and Hepatitis C Virus: The Troublesome Duo
The two hepatitis viruses are well-known causes of hepatocellular carcinoma (Donato et al., 2002; Hsu et al., 1988). Hepatocellular carcinoma is reported to be the fifth most common cancer globally and the third leading in terms of cancer deaths (De Martel et al., 2012). Infection with both hepatitis viruses is chronic and can occur together with inflammation (Takeda et al., 2017; Zeremski et al., 2007). Destruction of liver tissue can occur and this leads to regeneration and “wound healing,” ultimately resulting in scarring (Cordero-Espinoza and Huch, 2018; Wallace et al., 2008). Fibrosis can eventually evolve into hepatocellular carcinoma.
Molecular mechanisms involved in viral carcinogenesis
The development of mutations in the liver caused by long-lasting oxidative damage leads to the development of hepatocarcinoma. Direct involvement of hepatitis B virus (HBV) and hepatitis C virus (HCV) in the development of hepatocarcinoma has been recorded. For example, host gene expression is significantly changed through the actions of X antigen, which is encoded by HBV (Feitelson et al., 2014; Yan et al., 2009).
In addition, nonstructural protein 5A and nonstructural protein 3, both encoded by HCV, also change host gene expression (Chung et al., 1997; Davidson, 2009; Dolganiuc et al., 2003; Guo et al., 2018; Tellinghuisen and Rice, 2002). Liver cell proliferation, promoted by NS5A and NS3 through the activation of various signal transduction pathways including Wnt-B-catenin and affect cell survival, proliferation, and migration (Jeong et al., 2012). Of importance, NS3 and NS5A have been shown to promote cyclins and cdk2 expression. Inflammation caused by immune cells responding to the presence of viruses release large amounts of cytokines can promote tumorigenesis (Bréchot, 2004; Castello et al., 2010).
Several cancer hallmarks have been identified during viral-mediated progression to hepatocarcinoma. In the case of HBV infection, chronic hepatitis develops first followed by the development of fibrosis, cirrhosis leading to hepatocarcinoma. Early hallmarks after HBV infection include genomic instability and mutations. In addition, avoidance of immune detection, increased proliferation, evasion of growth suppressors, and replicative immortality are some of the extra early hallmarks. Cellular proliferation coupled to viral replication can result in extensive liver damage.
HBV-mediated cell cycling through the action of X antigen is known to activate several signaling pathways including PI3-Akt, NF-κβ, and Wnt signaling (Benhenda et al., 2009; Martin-Vilchez et al., 2011). By regulating transcription factors and micro-RNA, HBV-encoded X antigen influences host gene expression and introduces epigenetic changes (Tian et al., 2013; Yip et al., 2011). At later stages of hepatocarcinoma development, induction of angiogenesis, metastatic abilities, and inflammation is observed. X antigen is known to activate the transcription of hypoxia-inducible factor 1α (HIF-1α), leading to the upregulation of vascular endothelial growth factor expression for example, thus promoting angiogenesis (Martin-Vilchez et al., 2011).
In addition, X antigen has been shown to induce the expression and activation of matrix metalloproteinases (MMPs), enzymes needed to degrade extracellular matrix proteins and promote formation of new blood vessels as well as metastasis (Chung et al., 2004). HCV causes ROS generation, leading to the activation of many signaling cascades involved in the stabilization of HIF-1α (Ivanov et al., 2013; Mahmoudvand et al., 2019). Infection by these two viruses has been shown to cause the upregulation of cytokines involved in angiogenesis (Vrancken et al., 2012).
Both viruses are known to block apoptosis and thereby prevent growth suppression and immune detection. HBV-encoded X antigen, for example, prevents the activation of antiviral signaling protein needed for innate antiviral signaling (Feitelson et al., 2009; Tsunematsu et al., 2017). X antigen also blocks caspases-3 and -8 and thus prevents apoptosis and converts transforming growth factor-β (TGF-β) signaling into tumor promoting (Hussain et al., 2007; Staib et al., 2003). Hepatocytes expressing HBV are able to survive and continue growing through avoiding immune detection (Wang et al., 1991). HBV can act in a positive and negative way against apoptosis. HBV-encoded X antigen is known to promote phosphorylation of retinoblastoma and therefore inactivates it (Zemel et al., 2011). Furthermore, X antigen is also known to reduce the expression of many cyclin-dependent kinase (CDK) inhibitors (Zemel et al., 2011).
On the contrary, HBV-encoded X antigen can promote apoptosis in response to unregulated growth signaling that is activated by HBV-encoded X antigen (Feitelson et al., 2005). HCV-associated proteins can block apoptosis through inhibition of caspase-8. In addition, NS5A can adhere to signaling molecules and reduce apoptosis activation (Zemel et al., 2011). By being integrated near the telomerase reverse transcriptase gene, HBV-encoded X antigen causes overexpression of the gene, leading to replicative immortality (Feitelson and Lee, 2007). Human hepatocytes transfected with HCV core show increased expression of telomerase and dedifferentiated, and similar to HBV, leads to replicative immortality (Ray et al., 2000).
The upregulation of MMPs, receptors including hepatocyte growth factor receptor and c-met has been suggested to promote angiogenesis, migration, and metastasis associated with hepatocellular carcinoma (Block et al., 2003).
For example, HBV-encoded X antigen has been shown to enhance c-met expression (Xie et al., 2010). By enhancing the expression of MMPs, X antigen therefore promotes cell migration and invasive behavior (Zemel et al., 2011). HCV has been shown to inhibit TGF-β effects on tumor growth inhibition and promotes tumor migration and invasion (Caja et al., 2018; Chen et al., 2016). Chronic liver disease has been shown to be associated with regeneration, during which HBV genomic material is integrated into the host DNA. Integration of HBV DNA often occurs in region associated with cancer and those that are genetically unstable (Feitelson and Lee, 2007). In most cases HBV DNA integration occurs on the human telomerase reverse transcriptase (hTERT) gene with several integration genome encoding the HBV-encoded X antigen (Kew, 2011).
The inactivation of p53 by X antigen and inhibition of DNA repair proteins leads to genomic instability, making X antigen a key player in compromising genomic integrity (Martin-Vilchez et al., 2011; Matsuda and Ichida, 2009). HBV-infected cells, with a functional X antigen, are able to evade apoptosis by the involvement of X antigen in the stabilization of poly (ADP-ribose) polymerase (PARP). PARP is central in the protection of cells from apoptosis. HCV-associated protein NS5A also prevents apoptosis through stabilization of PARP-1 (Kuchay et al., 2018). The two viruses are known to affect cellular metabolism allowing metabolic switching in order for cells to grow and proliferate unchecked (Brault et al., 2013; Martinez-Pastor et al., 2013). Tian et al. (2013) demonstrated that both HCV and HBV enhance the expression of DNA methyltransferases that can inhibit tumor suppressor expression in hepatocellular carcinoma.
KSHV Carcinogenesis
The causal agent for Kaposi's sarcoma is the human herpesvirus-8 or KHSV (Cavallin et al., 2014; Dittmer and Damania, 2016). In Kaposi's sarcoma, spindle cells of either endothelial of vascular origin that are infected with the KSHV proliferate profusely and this is coupled to enhanced angiogenesis and infiltration of immune cells (Ganem, 2010; Mesri et al., 2010). It is important to note that KSHV is able to infect several cells including immune cells such as monocytes and B cells. Many of the oncoproteins encoded by KSHV can regulate cellular proliferation, survival, and evasion of the immune (Coscoy, 2007; Ganem, 2010; Mesri et al., 2010). The latent or lytic stage of KSHV involves the replication of the virus along with the host through the expression of the nuclear antigen, allowing the KSHV episome to adhere to the host chromosome (Mui et al., 2017). By adhering to the host chromosome, the KSHV episome is able to segregate during cellular division.
The lytic phase of the KSHV involves the expression of the viral genome, allowing the virus to replicate and be able to be infectious upon cell lysis (Ganem, 2010; Mesri et al., 2010). The latent and lytic phases of the KSHV are important for its development. Most Kaposi's sarcoma lesions contain mostly cells infected with the virus at the latent phase although cells are also present that are infected by the virus at the lytic phase (Ganem, 2010; Mesri et al., 2010).
KSHV infection and carcinogenesis
The latent phase of the KSHV includes transcripts that drive host cell proliferation and virus replication (Friborg et al., 1999; Mesri et al., 2014). Antiproliferation signals are blocked through the action of nuclear antigen, which blocks p53 and phosphorylated retinoblastoma pathways that are tumor suppressor pathways (Cirone, 2018; Herrera et al., 2005). Uncontrolled infected host cell proliferation also contributes to loss of genetic integrity (Si and Robertson, 2006). Downregulation of p27, a CDK inhibitor, is also associated with KSHV infection leading to increased host cell proliferation (Aneja and Yuan, 2017; Cavallin et al., 2014; Purushothaman et al., 2015).
MYC expression and therefore host cell proliferation is also enhanced by KSHV infection through the action of the KSHV nuclear antigen that stabilizes B-catenin (Fujimuro et al., 2003). Host cells infected by latent KSHV can avoid apoptosis through induction of the anti-apoptotic NF-κβ signaling (Guasparri et al., 2004).
It has been reported that micro-RNAs contribute to the activation of the NF-κβ signaling (Zhu et al., 2013). Overall, KSHV latently infected cells are able to proliferate indefinitely and avoid apoptosis. Furthermore the KSHV nuclear antigen is able to promote angiogenesis through stabilization of HIF-1α (Cai et al., 2006a). Several reports also show that NF-κβ signaling causes the release of proangiogenic cytokines including IL-6 (Grossmann et al., 2006). Some reports show that cells infected with the KSHV imitate cancer cells in terms of their metabolism (Delgado et al., 2012; Sanchez et al., 2015). KSHV infection causes a reduction in oxygen dependence and induces endothelial cells to lactic acid production (Delgado et al., 2010, 2012). A cytokine storm activated by the NF-κβ cascade results in inflammation observed in Kaposi's sarcoma pathogenesis (Grossmann et al., 2006; McCormick and Ganem, 2005). Activation of NF-κβ and the subsequent increase in Bcl-2 members helps latently infected cells to escape immune detection (Guasparri et al., 2004).
In order for KSHV to reproduce and make virions during the lytic phase of infection, the virus expresses its full replication program including viral Bcl-2 and G protein-coupled receptor to mimic the host's signaling regulators (Dai et al., 2018; Purushothaman et al., 2016). Several host cellular programs are affected including metabolism, immune evasion, and DNA damage control mechanisms. Several other KSHV products also partake in the recruitment of endothelial cells and thus promote angiogenesis and inflammation.
Several viral gene products, homologs of host cell gene products induce cellular signaling involved in promoting cellular proliferation, evasion of apoptosis, genomic instability, and insensitivity to antigrowth factors (Choi and Nicholas, 2010; Nakamura et al., 2001; Offermann, 2007). In addition, several viral genes associated with the lytic phase can also cause enhanced angiogenesis in infected cells (Bais et al., 2003), immortalization of infected endothelial cells through the increase in length of the telomeres (Wang et al., 2006) and metabolic reprogramming through regulation of tyrosine kinases (Karki et al., 2011), and enhanced inflammation (Brinkmann et al., 2007).
KSHV has been associated with cancer in individuals infected with HIV, immunocompromised individuals, individuals from regions such as Sub-Saharan Africa (SSA) as well as the elderly individuals from regions such as the Mediterranean (Ganem, 2010; Mesri et al., 2010). In individuals with HIV infection, the enhanced KSHV lytic reaction and promotion of angiogenesis is some of the factors associated with Kaposi's sarcoma development (Aoki and Tosato, 2007). In individuals who are immunocompromised, immune cells cannot remove KSHV-infected cells and this provides enough time for the KSHV to express its genes linked to increased angiogenesis.
Outlook
Preventing and clinical targeting of viral oncogenesis
Cancers associated with viruses can be treated using vaccines. Besides targeting host genes influenced by the presence of the virus, viral genes can also be targeted. At present, several combinations of therapeutic agents against viruses are in use for virus-linked cancers. Sorafenib is used to target vascular endothelial growth factor receptor 2 and platelet-derived growth factor receptor (PDGFR), for example, in HBV- and HCV-linked hepatocellular carcinoma. Ribavirin and protease inhibitors are used to target viral replication and protein translation in hepatocellular carcinoma as well. Rapamycin targets mTORC1 in AIDS-associated and transplant-linked Kaposi's sarcoma. In addition, Imatinib is used to target PDGFR and c-kit in AIDS-associated Kaposi's sarcoma. Cancers that are associated with viruses are still a challenge especially given that there are currently no models to use during investigations.
Treatment of EBV-associated malignancies
Both EBV- and HPV-associated malignancies are treated through chemotherapy and radiotherapy (Méndez-Vilas, 2013). Recent advances in the treatment of both EBV- and HPV-linked malignancies include the use of cell therapies and several antibodies that targets EBV- and HPV-associated oncoproteins. Cells that are infected with EBV can also be destroyed by induction of toxic genes expression and the induction of lytic gene expression in cells infected by the EBV virus (Frappier, 2012; Israel and Kenney, 2003; Westphal et al., 2000).
Several therapies including the use of radiation, DNA methyltransferases, and chemicals such as sodium butyrate have shown promise in the treatment of EBV-associated malignancies (Mentzer et al., 2001; Westphal et al., 2000). Commonly used drugs such as cisplatin, 5-fluorouracil, and doxorubicin have also demonstrated the ability to induce EBV lytic replication with further studies underway to determine the required doses (Liu and Mamorska-Dyga, 2017). A combination of different therapies through the use of both lytic gene expression induction and targeting of tumor cells infected with EBV can also be used successfully (Liu and Mamorska-Dyga, 2017).
One major problem associated with lytic EBV induction is the possibility of enhanced viral dissemination and the possible development of lymphomas linked to EBV (Kanakry and Ambinder, 2013). Several antivirals such as acyclovir and ganciclovir have shown the ability to reduce viral replication but suffer the lack of effectiveness during the latent phase of EBV replication (Hutajulu et al., 2014). It is very difficult to target EBV at the latent stage of infection as EBV makes use of the host replication system and host cells will likely be affected by any therapy. Latent proteins can however be targeted and therapies are still under development (Li et al., 2018).
For example, hydroxyurea can target EBV episome (Liu and Mamorska-Dyga, 2017). Furthermore, new therapies such as cidofovir act against EBV by interfering with the function of its latent proteins (Kanakry and Ambinder, 2013). Syk inhibitors including entospletinib and fostamatinib have been developed and these block the syk signaling necessary for B cell survival (Frappier, 2012). Inhibitors of PI3K-Akt signaling such as LY294002 and rapamycin can stop tumor growth in EBV-associated gastric cancer and Burkitt's lymphoma, respectively (Cen and Longnecker, 2011; Shin et al., 2010).
Besides inhibitors, monoclonal antibodies and vaccines can also be used for the treatment and prevention of EBV-linked diseases. For example, rituximab is a monoclonal antibody used in the treatment of post-transplant lymphoproliferative disorders, a disease associated with transplantation of solid organs including kidney, lung, heart, and liver (Kanakry and Ambinder, 2013; Kanakry et al., 2013). In addition, the targeting of CD70, expressed by cells infected by EBV, by monoclonal antibodies has been shown to reduce cell proliferation (Kanakry and Ambinder, 2013; Kanakry et al., 2013). Several reports show that vaccinations can induce viral immunity in animal studies and humans (Liu and Mamorska-Dyga, 2017).
Treatment of HPV-, HBV-, and HCV-associated malignancies
Vaccines that can prevent HPV infection are readily available today including Cervarix that is specific for the high-risk HPV type 16 and 18 (Signorelli et al., 2017). Vaccination with HPV vaccines can be for most HPV types and usable by both male and female patients (Cuzick, 2015). Most HPV vaccines are made from recombinant L1 major capsid protein but are known to induce an immune reaction as well (Harper and DeMars, 2017; Panatto et al., 2015). HPV vaccines based on the L2-minor capsid protein have so far demonstrated broad yet less efficacy compared with L1-based vaccines (Campo and Roden, 2010; Schiller et al., 2012). Cytotoxic and helper T cells induced by HPV vaccines are able to target human cells expressing the E6 and E7 oncoproteins (Kim and Kim, 2017; Nakagawa et al., 1997).
Viral replication suppression and therefore abrogation of hepatocellular carcinoma progression is the main goal of HBV treatment. Therapy for the rest of life can greatly reduce fatalities and prevent relapse of hepatocellular carcinoma (Cheng et al., 2009; Niu and Hann, 2017). A lot of hepatocellular carcinoma cases are found in Asia-Pacific region and many suffer from chronic hepatitis B infection (Cheng et al., 2009). Studies have shown that sorafenib can be effective at treatment of advanced hepatocellular carcinoma (Cheng et al., 2009; Kudo et al., 2011). Treatment is based on the amount of HBV DNA detected and the presence of fibrosis and cirrhosis (European Association for the Study of the Liver, 2017a; Sarin et al., 2016). Other forms of treatment include drugs such as pegylated IFN-α as well as adefovir and telbivudine (Terrault et al., 2016).
Chronic hepatitis caused by HCV leads to hepatocellular carcinoma. In most cases treatment for hepatocellular carcinoma caused by HCV is similar to that of HBV-induced hepatocellular carcinoma. Successful treatment as evidenced by sustained virological response leads to undetectable levels of HCV RNA for a minimum of 3 months after treatment (Tilly et al., 2015). Patients with advanced fibrosis and cirrhosis are at higher risk of complications that can result in death and are usually prioritized during treatment (European Association for the Study of the Liver, 2017b).
IFN-α acts by inhibiting cellular processes induced by HCV infection such as angiogenesis and fibrogenesis (Khan et al., 2008; Zamor et al., 2017). Antiviral agents demonstrate less adverse effects and high efficacies of >50%, resulting in them being used as first-line therapies to treat HCV infection (Zamor et al., 2017). In many patients, hepatic function is greatly improved after treatment with antiviral agents. Many low- to middle-income countries cannot readily provide antiviral agents as they are costly and many patients with cirrhosis may require a longer time of treatment (Serti et al., 2017; Zeuzem et al., 2009).
Conclusion
Overall, cancers caused by viruses are a result of the viruses adapting and developing strategies to survive and replicate within a host cell via activation of several oncogenic mechanisms linked to immune evasion and angiogenesis. As many viruses require long periods of incubation time before causing cancers, this provides an opportunity to prevent and treat such cancers. Research linking viruses to cancers is increasing every day and novel therapeutic agents are under development and trial for either eradicating viruses, suppressing viral replication and destroying virus-infected tumor cells within the tumor microenvironment. One appealing strategy on targeting viruses implicated in tumorigenesis is the fact that most viruses affect similar signaling cascades during tumorigenesis. Thus, studies and drugs that act on one virus may be applied to all viruses implicated in different cancers.
Footnotes
Author Disclosure Statement
The author declares there are no conflicting financial interests.
Funding Information
No funding was received for this article.