
Abstract
MicroRNAs (miRNAs) that are mutually modulated by their interacting partners (interactome) are being increasingly noted for their significant role in pathogenesis and treatment of various human cancers. Recently, miRNA interactome dissected with multiomics approaches has been the subject of focus since individual tools or methods failed to provide the necessary comprehensive clues on the complete interactome. Even though single-omics technologies such as proteomics can uncover part of the interactome, the biological and clinical understanding still remain incomplete. In this study, we present an expert review of studies involving multiomics approaches to identification of miRNA interactome and its application in mechanistic characterization, classification, and therapeutic target identification in a variety of cancers, and with a focus on proteomics. We also discuss individual or multiple miRNA-based interactome identification in various pathological conditions of relevance to clinical medicine. Various new single-omics methods that can be integrated into multiomics cancer research and the computational approaches to analyze and predict miRNA interactome are also highlighted in this review. In all, we contextulize the power of multiomics approaches and the importance of the miRNA interactome to achieve the vision and practice of predictive, preventive, and personalized medicine in cancer research and clinical oncology.
Introduction
There has been a significant increase in the number of studies on the role of miRNAs in normal physiological processes and diseases, including cancer (Peng and Croce, 2016). Both individual and group of miRNAs are known to regulate the progression of carcinogenesis, metastasis, and tumor microenvironment (Zaheer et al., 2019). MiRNAs targeting oncogenes are termed as tumor suppressor miRNAs, and those targeting tumor suppressors are termed as oncomiRNAs (Svoronos et al., 2016). They are being increasingly used as biomarkers (Isenovic et al., 2017), and in miRNA replacement or inhibition therapies (Hosseinahli et al., 2018; Mollaei et al., 2019; Nguyen and Chang, 2018). MiRNAs play an important role in the therapeutic resistance of cancers and many are currently in various phases of clinical trials (Guo et al., 2020; Johora et al., 2019; Ors-kumoglu et al., 2019; Si et al., 2019).
The growing importance of miRNAs in cancer research and personalized medicine (Akyay et al., 2020) led to investigation of miRNA interactome using single omics followed by multiomics. Multiomics enables identification/characterization of biomarkers, disease progression, and treatment response, and more recently is being used to characterize miRNA interactome. This review elaborates on various studies involving multiomics approaches in identifying miRNA interactome using cancer models, with special mention on proteomics technologies and the new single-omics technologies. It also discusses the upcoming challenges, possible solutions, and prospects. Since exosomal and circulatory miRNAs and their roles in cancer are covered in several reviews independently (Bhome et al., 2018; Cui et al., 2019; Kamal and Shahidan, 2020; Sun et al., 2018; Wang et al., 2018), they are excluded in this review.
MiRNA Interactome
MicroRNAs (miRNAs) are small noncoding RNAs (18–22 nucleotides) discovered in Caenorhabditis elegans (Lee et al., 1993) and their biogenesis involves initial transcription as pri-miRNAs, processing by Drosha/DGCR8 (DiGeorge syndrome critical region 8) enzyme complex to pre-miRNAs in the nucleus, later exported by exportin-5, and finally processed into mature miRNAs by the enzyme dicer 1 in the cytoplasm (Denli et al., 2004; Han et al., 2004; Lee et al., 2003; Zhang et al., 2004). Mature miRNA strands are then loaded into RNA-induced silencing complex (RISC) to target the specific protein-coding messenger RNAs (mRNAs). Based on the complementarities between miRNAs and mRNAs, the mRNAs are either degraded (completely or partially) or translationally inhibited (Brien et al., 2018; Kawamata and Tomari, 2010).
MiRNAs not only interact with their target mRNAs but also partner with long noncoding RNAs (lncRNAs, >200 nucleotides in length) and pseudogenes (nonfunctional genes), through competitive binding, resulting in the disruption of several signaling pathways such as angiogenesis and stem cell development (López-urrutia et al., 2019; Sen et al., 2014). In fact, they form a vast regulatory network interacting with the promoter/enhancer regions and transcription factors of target/host genes and modulate gene expression negatively in most cases or even positively in some cases (Tan et al., 2019). MiRNAs also interact with RNA binding proteins (RBPs), including Drosha and Dicer 1 and other RBPs specific to a given miRNA, and inhibit their binding to argonaute 2 (AGO2) protein.
Alternatively, miRNAs could modulate the RBP genes and contribute to the diversity in physiological and pathological processes (Chen et al., 2019; Ciafrè and Galardi, 2013; Zealy et al., 2017). Recent studies have highlighted the importance of polymorphisms in miRNAs and their interacting partners in physiology and pathology (Hrovatin and Kunej, 2018; Piletic and Kunej, 2018).
Figure 1 depicts the miRNA interactome and these interactions can be co-operative or competitive in nature, for example, PUM1 promotes stronger binding of miR-221 and miR-222 to CDKN1B 3′ UTR, promoting its degradation (Kedde et al., 2010).

MiRNA interactome: the figure depicts miRNA interacting partners and style of interaction whether miRNAs can act co-operatively or competitively with their partners.
Bioinformatics and Experimental Tools to Identify miRNA Interactome
There are many bioinformatics tools to predict the targets for miRNAs (Ab Mutalib et al., 2019). However, they predict targets based on seed sequence complementarity, free energy, and site conservation status, which are insufficient to predict the total interacting partners of miRNAs as they often end up with significant false positives and/or false negatives; therefore, experimental validation is necessary and this can be accumulated only through single-omics experiments.
MiRNA-mRNA pairs have been identified using reporter assays, cross-linking immunoprecipitation (CLIP), affinity pull-down, degradome sequencing, or bioinformatics prediction (German et al., 2008; Hafner et al., 2010; Helwak et al., 2013; Keene et al., 2006; Kern et al., 2019; Licatalosi et al., 2008; Lu and Leslie, 2016; Moore et al., 2015; Willmann et al., 2014; Yoon et al., 2012). For a specific miRNA, it can be achieved by gene expression profiling after overexpression or inhibition, immunoprecipitation (IP) of RISC with a specific antibody, and pull-down with labeled miRNA mimics (Li and Zhang, 2019).
MiRNA interaction with lncRNAs is identified using CLIP (Kazimierczyk et al., 2020) and enhanced cross-linking and immunoprecipitation (eCLIP), which can additionally identify RBPs regulating miRNAs (Nussbacher and Yeo, 2018). Ribosome profiling is another method that identifies miRNAs associated with ribosomes and unravels changes in protein levels of miRNA targets mediated by translational repression or mRNA degradation and their regulators (Bazzini et al., 2012; Cottrell et al., 2017; Eichhorn et al., 2010).
Expression profiles of miRNAs can be quantified using microarray (Li and Zhang, 2019). Pull-down assays utilize biotinylated miRNAs to identify their partners being pulled down using streptavidin beads and later sequenced. IP uses antibodies against proteins in RISC, particularly argonaute proteins (AGO), to identify mRNAs bound to a miRNA. Modifications include photo-crosslinking of bound RNAs with AGO, such as in CLIP or photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) and cross-linking, linking, and sequencing of hybrids (CLASH is a modification of CLIP method, including sequencing of hybrids) (Li and Zhang, 2019). Degradome sequencing identifies miRNA cleavage sites leading to novel target identification. Figure 2 shows the various tools to identify miRNA interactome and the type of interaction detected by these tools.
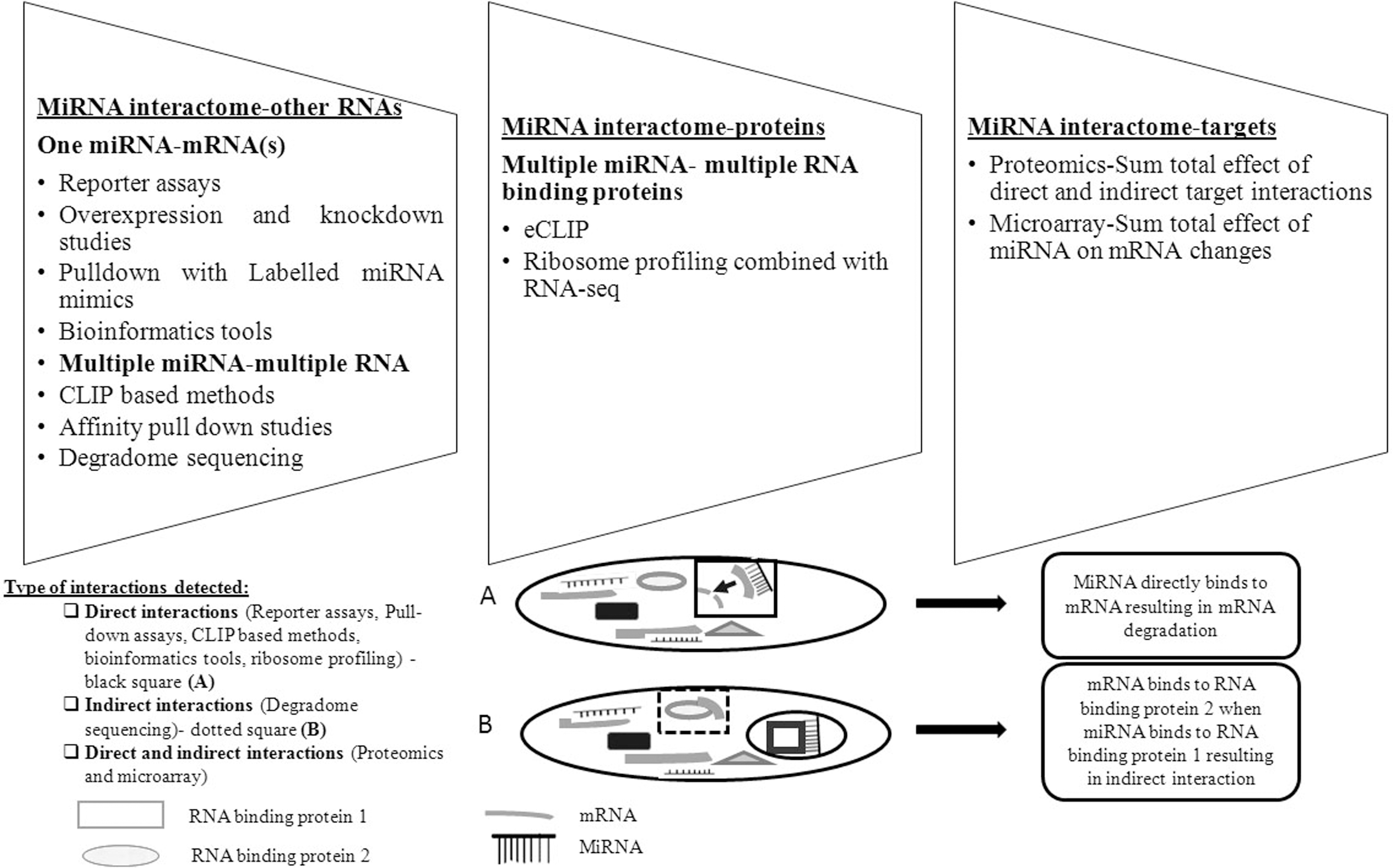
Tools to characterize miRNA interactome: the figure enlists the various methodologies and techniques used to identify each aspect of the interactome and the type of interactions detected by these tools. The inset A shows direct interaction of miRNA to target leading to its degradation (in black square) as indicated by the arrow, while inset B shows how miRNA directly interacts with RBP 1 (black circle), but the free mRNA binds to other RBP 2 (in dotted square) in the cell, making this RBP 2 an indirect interacting partner of miRNA and alters the cellular response.
Microarray reflects only on the changes in the target mRNAs from a single or multiple miRNA(s), but miss out on changes mediated by translational repression. Similarly, other tools like IP, CLIP, and its associated modifications identify direct interactions between miRNAs, isomiRNAs (isomiRs are variations of miRNAs that are generated from a single miRNA precursor locus), and their partners, that is, they identify targets regulated by both translational repression and mRNA degradation, but leave out the indirect interactions. The results from these experimental studies provide the basis for the single-omics data, that is, global measurement of the subset of cellular processes or molecules.
Single Omics as a Tool to Study miRNA Interactome
Single omics have long been used to identify biomarkers, differentially expressed genes, proteins, miRNAs, and miRNA targets to distinguish between different physiological and pathological states of the cell. Genomics delves into the organism's genome, particularly the structure, function, and mapping of genes, whereas epigenomics deals with changes in the DNA without changes in its sequence such as associated histone modifications and DNA methylations. Transcriptomics exhibits the transcriptional status of the cell at specific circumstances, including protein-coding mRNAs and noncoding RNAs and their associated properties. Proteomics reveals information about the various cellular protein levels and their post-translational modifications, and metabolomics divulges details about the small molecules called metabolites derived from cellular processes.
Individual omics studies only one part of the miRNA interactome, such as genomics would reveal changes in the miRNA gene that affect its expression; epigenomics would reveal epigenetic factors influencing miRNA expression or vice versa; transcriptomics would reveal the overall RNA targetome of the miRNA interactome; and unlike the rest, proteomics would quantify miRNA-induced changes at the protein level studying both direct and indirect interactions.
Many techniques have been developed to label and quantify proteins (Giambruno et al., 2018). Various techniques involving mass spectrometry (MS)-based proteomics, with modifications such as affinity purification, pull-down assays, and hybridization-based purification, are known (Giambruno et al., 2018). Untargeted proteomics involves covering as much of the proteome as possible, while targeted proteomics aims to target a specific subset of the proteome to improve specificity and sensitivity, which can be used for developing biomarkers and to unravel the details on the targetome of miRNA(s) (Nouri-Nigjeh et al., 2015).
To further improve single-omics methods to detect and quantify miRNAs, several developments have been made and shown in Table 1. The advancements in single omics are mainly an improvement over quantitative real-time PCR (qRT-PCR) to detect and quantify miRNAs. Validation of omics from microarray is done using qRT-PCR and although it is quite sensitive, there are still some disadvantages such as lack of specificity and inability to distinguish between genome location of miRNA host genes and their paralogs, and is labor- and cost-intensive (Forero et al., 2019). Lack of specificity and labor-intensiveness has been overcome by MS-based proteomics (Xu et al., 2015) and it converts miRNA signal into reporter peptide signal, which is later quantified by MS using a peptide substrate that is complementary to target miRNA.
Single Omics Advances in the Context of Cancers
N/A, not applicable.
Another tool that pointed out errors in the annotation of dominant miRNA strands using an advanced sequencing method is AQ-Seq (Kim et al., 2019). It overcomes the bias in conventional methods and helps to identify miRNAs with correct 5’ end, their isomiRs, and modifications, all of which are important in target prediction and some of them can predominantly distinguish tumor from normal tissues across 32 TCGA cancer types (Loher et al., 2014; Telonis et al., 2015, 2017). This can probably explain the observed discrepancies between miRNA and target levels in real experiments due to differential targeting by isomiRs and help to quantify miRNAs and isomiRs accurately, and better understand how these modifications affect their targets when combined with other omics.
A recent study highlighted a method to identify spatially resolved and multiplexed miRNA tissue array to determine the spatial distribution of miRNAs in tumor tissues (Ho and Lee, 2015; Nagarajan et al., 2020; Turunen et al., 2019). The array has wells with reagents that can release miRNAs from the tissue, and then probes polymerized in hydrogel can quantify the released miRNAs. This can open new avenues to visualize tumors better and predict disease prognosis and treatment response in complex samples (Nagarajan et al., 2020).
To overcome the difficulties with commonly used MS-based proteomics methods such as extensive labeling protocol, high costs, and limitations in detection of complete proteome, a novel method was developed utilizing a new O16/O18 labeling method combined with other approaches and statistical tests to identify truly significant differentially expressed proteins and recognize the strongly positive targets of miRNAs of interest from them (Ma et al., 2015).
In continuation with the above developments, a novel tool multiple reaction monitoring (MRM)-ion pair finder was developed to combine the advantages of targeted and untargeted approaches for protein selection and quantification and sufficiently reduce time on manual selection of appropriate ions for quantitation as in targeted method. It helps to identify both known and unknown metabolites from real biological samples with significant accuracy and reliability and reduced false positives (Luo et al., 2015).
The above developments allow an indirect quantification of miRNAs without the hassles of amplification and use of fluorophores and can be expanded for a panel of miRNAs to be used as prognostic or diagnostic markers in a clinical setting. Several such indirect quantifications developed earlier can also be used to detect and/or quantify a single miRNA or a panel of miRNAs by amplification or a unique probe readout, which have possible limitations as qRT-PCR, warranting further processing and refinement (Cadoni et al., 2020; Cheng et al., 2018; Dave et al., 2019).
Overall, these improved and advanced omics technologies can increase the reliability of data obtained and if integrated together with other omics can provide a significant knowledge base for miRNA interactome and its undiscovered characteristics. They can be applied in the final step of multiomics study for a given phenotype or in intermediate steps to generate an inclusive list of biomarkers.
Multiomics as a Tool to Study miRNA Interactome
Multiomics approach includes a combination of single-omics technologies such as next-generation sequencing and MS for genomics, epigenomics, transcriptomics, proteomics, and metabolomics (Aebersold and Mann, 2003; Dettmer et al., 2007; Kulski, 2015), which gives a unique set of data with its advantages and disadvantages. The wide range of molecules interacting with a miRNA makes it evident that the integration of multiple data types is necessary to obtain a holistic picture of miRNA interactome. The significance of multiomics in providing a better understanding of diseases by extracting the most from a single layer of data of different types and analyses of various strategies to understand disease-causing agent has been studied (Hasin et al., 2017).
Use of different omics approaches combined with bioinformatics for predictive, preventive, and personalized medicines (PPPMs) has been reported in cancers (Yoo et al., 2018). Combined or integrated omics, such as proteogenomics and proteometabolomics, is currently gaining momentum in various cancers to identify biomarkers for PPPM and can accelerate the detection and prevention of cancer. Different algorithms are used in integrating multiomics to give meaningful information that might help in clinical applications (Huang et al., 2017).
However, these methods include one or a few omics, particularly gene expression, miRNA expression, copy number variations (CNV), and methylation status and miss out on the other omics. In addition, there is a lack of methods to integrate data from multiple cohorts, which is the demand of the hour to generate useful clinical data. Employing large number of subjects for analysis to gain meaningful insight from multiomics data is a limitation, but the increasing availability of large-scale human studies like Metabolic Syndrome in Man (METSIM) (Laakso et al., 2017) and Genotype Tissue Expression (GTEx) (Aguet et al., 2017) mitigates this. In this direction, Chakraborty et al. (2018) reviewed those reports involving a minimum of two or more omics, mostly the cancer repository: the cancer genome atlas (TCGA) (Weinstein et al., 2013) data.
Several interesting observations came out of multiomics, such as a) diversified applications of radiomics, a bridge between imaging and personalized medicine, with other omics and b) the use of system biology to develop strategies for personalized medicine (Lu and Zhan, 2018; Yoo et al., 2018). A common observation (Chakraborty et al., 2018; Yoo et al., 2018) is that single omics cannot indicate the causative relationship between the observed signatures and the disease, highlighting the advantage of multiomics over single omics.
Taken together, these reports indicate a significant understanding of multiomics in cancer prevention, treatment, and prediction of disease progression with a lack of discussion on miRNA interactome involving multiomics in cancer. Thus, in this review, we attempted to bridge this gap by focusing on two key aspects:
Role of multiomics in studying miRNA interacting partners with various approaches and bioinformatics tools The future applications of multiomics in miRNA and conventional therapy combination approach and treatment response optimization.
Figure 3 illustrates various outcomes of integration of multiomics for miRNA interactome characterization. An interactome network between miRNAs and targets can showcase molecular interactions in the form of nodes (units of the network) and critical hubs (nodes with the greater number of interactions than average) with diagnostic or prognostic potential.
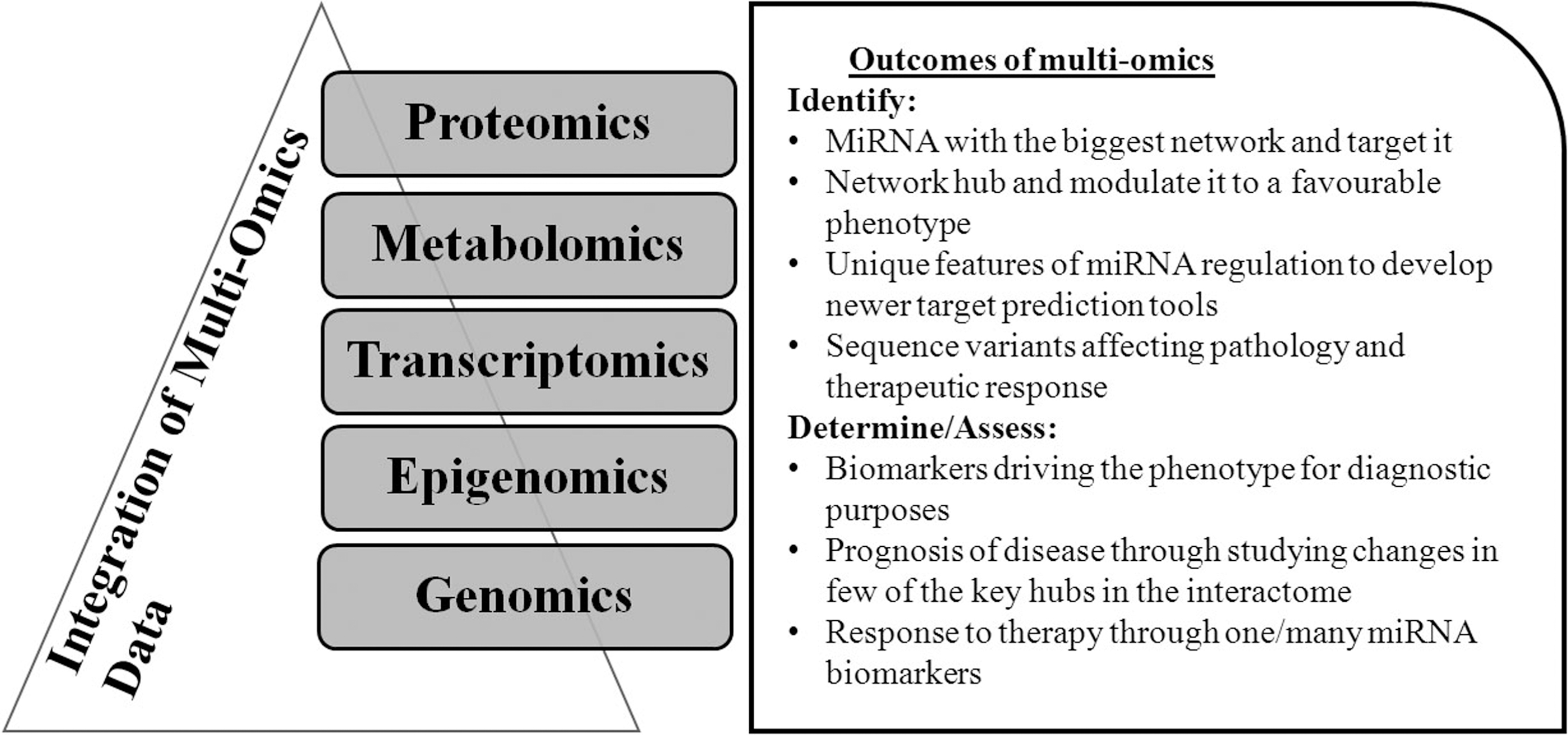
Multiomics in miRNA interactome and its possible outcomes.
Application of Multiomics in miRNA Interactome Identification
Two different approaches can be used to understand how a miRNA or multiple miRNAs work together during the progression of cancer and if they can act as prognostic/diagnostic markers and/or therapeutic targets, (1) a disease model with its phenotype and a pattern involving a miRNA or multiple miRNAs and (2) the effect of a miRNA or its cluster on its interactome.
A Disease Model-Based Interactome Identification
In this approach, the first step is to characterize different phenotypes modulated by miRNAs such as metastasis, downregulation of specific protein, therapy sensitivity, and others. A list of the studies discussing the disease model-based interactome identification, including omics and software used for analyses, is shown in Table 2.
Various Omics Platforms and Software Used for Multiomics Analyses
When DDX5 (DEAD-Box Helicase 5), an RNA helicase and noncoding RNA processing enzyme upregulated in breast cancer, was knocked down, 2 proteins (profilin and cofilin) were found to be upregulated (by proteomics) (Wang and Huan, 2012). Furthermore, miRNA profiling revealed DDX5 downregulation by a subset of miRNAs (miR-21, let-7 family, and miR-182), bioinformatics analysis of which revealed miRNA-182 to be responsible for the observed phenotype, providing a unique target for therapy against basal-type breast cancers.
In another study, the comparison of differentially expressed miRNAs from cell line studies (chronic exposure to cigarette smoke vs. parental) with TCGA datasets (smoker vs. nonsmoker) revealed overlapping miRNAs, confirming the in vitro study, following which combined miRNA profiling and proteomics analysis identified miRNA target pairs and pathways dysregulated in response to cigarette smoking, giving an integrated picture of the effect of cigarette smoking on cells (Advani et al., 2017).
Another study identified that glycolysis is commonly dysregulated in lung cancer and in disused diaphragm muscle by proteomics and used microarray to identify miRNA(s) regulating the rate-limiting step (Phosphofructokinase) in glycolysis, elucidating the mechanism of these disease phenotypes (Tang et al., 2012).
The combination of five omics data namely DNA copy number, DNA methylation, mRNA expression, miRNA expression, and reverse phase protein array (RPPA) was used to identify unique signatures in hepatocellular carcinoma (HCC) by TCGA group, to classify the subtypes in HCC: iclust 1, 2, and 3, with iclust 1 being the one with lower survival. This study has also identified different gene signatures that can be used to provide targeted therapy (Wheeler and Roberts, 2017). Furthermore, in this study, a minor class of HCC (iclust 1 subclass with isocitrate dehydrogenases (IDH) mutations), identified through integrative analyses of mutation and gene expression followed by miRNA expression, revealed miR-122 as a dysregulated miRNA for IDH mutant and IDH-like samples with promoter hypermethylation as the mechanism of dysregulation with its targets identified by miRNA-mRNA anticorrelation studies.
Together, this study has classified HCC subtypes, identified a minor subtype with poor prognosis, and a prospective target (miR-122) for the same subtype. This study highlights the advantages of multiomics studies emphasizing that the research integration from different reports can progressively develop therapeutic targets precisely than independent investigations.
In another investigation in colorectal cancer, identification of differentially expressed miRNAs has led to a group of 3 upregulated miRNAs (miR-424-3p, miR−503, and miR −1292) associated with overall survival and disease-free survival (Torres et al., 2018). These miRNAs share common targets identified by proteomics studies, while none of the bioinformatics tools predicted even one of them. This unique approach combining miRNA transcriptomics and proteomics gives a set of miRNA-target pairs that can be used as therapeutic targets to stall or prevent metastasis.
Another study in colorectal cancer elucidated the mechanisms of miRNA-mediated effects by combining miRNA and mRNA expression profiling with proteomics data (Liu et al., 2013) and revealed different mechanisms (mRNA Decay; mRNA Decay with other mechanisms; translational repression; translational repression with other mechanisms; mRNA decay; and translational repression, both strong or both weak) for miRNA regulation. Also, it highlighted the features (8mer site, site positioning within 3'UTR, local AU-rich context, and additional 3’ pairing) responsible for the same, which could be potentially used to develop newer computational target prediction tools.
Nassa et al. (2014) attempted to reveal the mechanism of a relatively less studied estrogen receptor β (ESR2) in breast cancer, which mediates estrogen-independent effects (Nassa et al., 2014). Although many dysregulated proteins were identified as targets for multiple miRNAs with miRNA profiling and proteomics profiling, the correlation between mRNA and miRNA was only moderate, indicating translational repression as a major mechanism. A subset validation of a single differentially expressed miRNA (miR-30a-5p) and its target mRNA (SNAI1), and protein levels found it to be correct. Molecular changes deduced between estrogen receptor-positive (ER+) and triple-negative (TNBC) breast cancer subtypes by utilizing differential miRNA and proteomics profiling and system biology approach revealed a differential single functional module of miRNAs and proteins (Berges-soria et al., 2015). This multiomics strategy involving system biology paves the way for envisaging the system in action and reveals novel therapeutic targets.
Reverse phase protein array (RPPA) is a low-throughput proteomics method based on antibody validation, but is still used when a specific phenotype is being addressed; an miRNA signature (miR-1, miR-133a, miR-133b, miR-135a, miR-143-3p, miR-145-3p, miR-205, miR-221-3p, miR-221-5p, miR-222-3p, miR-24-1-5p, and miR-31) was found to be suppressed in metastasis in multiple datasets, with relevance to prostate cancer (PC) (Coarfa et al., 2015).
Mechanisms of this dysregulation were identified by DNA methylation, copy number alteration signatures, and prognostic value through previously available datasets. Proteomics analysis of these miRNAs with RPPA revealed only a fraction of interactions of miRNAs with targets to be direct, reiterating the importance of understanding both direct and indirect effects of miRNAs as stated earlier.
Furthermore, Uhlmann et al. (2012) tried to overcome the limitations of RPPA such as antibody inaccuracy and utilized proteomics analysis to identify miRNAs regulating a single pathway. This led to the development of a low stringency network consisting of those targets bearing at least one 6mer evolutionary conserved site in 3’ UTR, showcasing the interactions of co-regulated proteins by one or more miRNAs (miR-124, miR-147, and miR-193a-3p) in pathway modulation.
An end to end study (Seviour et al., 2016) used a powerful iteration of clinical (TCGA) and in vitro data to determine the clinically relevant therapeutic targets for an RPPA-identified signature (CDKN1B, Myc, and phospho-Rb 1) in breast and ovarian cancers. Then, five corresponding miRNAs (miR-124, miR-365, miR-34b*, miR-18a, and miR-506) targeting this signature were identified from the proteomics data (TCGA and RPPA data of cell line models). The miRNA list was further refined to a single miRNA (miR-124) by validation on the other cancer models and strengthened by evidence in drug sensitization using the in vivo models. This report thus provides a holistic picture, resulting in a robust therapeutic target utilizing a plethora of data and omics.
Effects of miRNA and Its Clusters on Its Interactome
Another approach that uses miRNA-based proteomics to identify its interacting partners and mechanism of action has been reported (Giambruno et al., 2018) and several other reports where miRNA targets/interactome identified by proteomics are shown in Table 3.
MiRNA-Based Interactomes
N/A, not applicable.
A combination of proteomics and mRNA microarray was used to identify miR-34a targets (Chen et al., 2011) using isotope-coded affinity tags (ICAT) labeling and reverse phase liquid chromatography (RPLC)-MS/MS to identify novel targets with higher efficiency compared to matrix-assisted laser desorption ionization time of flight (MALDI TOF) MS/MS (Cheng et al., 2010). This shows that among proteomics techniques, each has its advantages and disadvantages, and therefore multiomics is a necessary tool to overcome the drawbacks of single proteomics techniques (Li et al., 2012).
An earlier study combined the proteomics and microarray analysis to show differential targets between an individual miRNA (miR-143 or miR-145) and its cluster (Bauer and Hummon, 2012). A similar study combined mRNA microarray and RPPA with prediction programs to identify direct targets of miR-200bc/429 to understand how the differential effects of 2 clusters of miRNA-200 family are mediated through their exclusive targets (Uhlmann et al., 2010). Although this is not a large-scale multiomics study, it succeeded in understanding the two different miRNA clusters of the same family differentially affecting the phenotype in the same cell line.
A miRNA signature from clinical samples followed with proteomics study of a single family (let-7 family) to identify the common targets of the family was reported (Kobayashi et al., 2014), similar to the strategy of disease model-based miRNA interactome identification. Unlike the Kobayashi study, two separate studies identified targets of miRNA-143 and miRNA-145 using proteomics, and they compared mRNA and miRNA changes to understand the mechanism of targeting by miRNA-143/145, that is, either mRNA degradation or translational repression (Huang et al., 2015; Yang et al., 2010).
These studies used a single miRNA proteomics approach that helped identify targets of single miRNA/family, but unlike the first disease model-based approach, it does not present us with a holistic view. However, this approach helps in enlisting the unique properties of miRNA regulation and its targets on a large scale. Most of the later studies focused on the disease instead of a single miRNA, to understand multiple dimensions of the disease.
Role of Multiomics and miRNA Interactome in Therapeutic Efficacy
Novel strategies such as combination therapy of tumor suppressor miRNAs with conventional therapy appear promising to overcome the resistance to conventional cancer therapies and prevent the recurrence of the disease (Ebrahimi and Hashemy, 2019; Mognato and Celotti, 2015). Studies with individual miRNAs predict sensitivity to therapy, while multiomics studies identify biomarkers that correlate with drug sensitivity or establish the mechanism of action of novel compounds.
A large-scale multiomics study utilized a combination of different datasets (mentioned in Table 4) and gave a pretty robust signature of genes, miRNAs, mutations, copy number alterations, and transcription factors, that is, biomarkers to predict responders and nonresponders (Bosquet et al., 2014). Another omics study of a combination of mRNA, miRNA microarray, and proteomics was utilized to understand the mechanism of action of a novel compound (shikonin) against leukemia (Wiench et al., 2013). Furthermore exploratory investigations on these studies will help identify those strongly modulating miRNAs that can be used as radiosensitizers or chemosensitizers. Immunotherapy is also being used in cancer treatment involving two major strategies: checkpoint inhibitors and adoptive cell therapy (tested in multiple cancer types like glioblastoma) (Matsiko, 2018).
MiRNA-Based Multiomics Data Applications to Predict Therapeutic Response
In a first of its kind multiomics study to hypothesize sensitization to immunotherapy, proteomics and miRNA microarray were used to identify the survival-associated biomarkers to determine response to tumor lysate-charged dendritic cell (DC) vaccination (Erhart et al., 2020). There is a dearth of literature in multiomics with miRNAs for therapy resistance, although many individual studies where a single miRNA or miRNA family such as miR-34 have been implicated in radiation response as well as their role in radiosensitizing cells (Chaudhry, 2013; Marta et al., 2015; Moertl et al., 2016).
Only one study showcases the predictive power of drug sensitivity in cancer cells by single-omics technologies, that is, proteomics methods, MS and RPPA, revealing that one could be better than other technologies for prediction of sensitivity, but multiomics components can complement each other and overcome shortcomings, while adding new information to the pool, drastically improving the predictive power (Ali et al., 2018). Multiomics can play a key role to understand the global response of cells to cytotoxic agents (radiation, drugs, or immune cells), to identify key signature molecules/targets/miRNAs in resistant tumors and in personalized treatment plans, to enhance response in resistant tumors. A summary of miRNA-based multiomics for therapy response as well as key cancer response pathways is depicted in Table 4.
Bioinformatics Tools to Predict miRNA Interactome from Proteomics Data
A list of tools (input/output data, programming language used, model used in development) that are available to identify miRNA targets by incorporating proteomics data as well as to identify its interactome is given in Table 5 (Li et al., 2009; Qin et al., 2013; Reczko et al., 2012). Some of the models used in these tools predict targets from input data either by probabilistic determination or machine learning. There is a lack of interest in developing applications for omics integration in miRNA target prediction, although there are several independent tools for analyzing quantitative proteomics data and miRNA target prediction. Since miRNAs exhibit unique regulation, newer methods are required to correctly dissect direct and indirect miRNA interactions with targets identified from proteomics.
Bioinformatics Tools to Forecast MiRNA Interactome from Proteomics
N/A, not applicable.
Future of Multiomics
Currently, most of the multiomics studies aim to identify prognostic and diagnostic markers (Kong et al., 2020; Kwon et al., 2015; Yang et al., 2019) and classify subgroups of different cancers (Aure et al., 2016; Zhang et al., 2016b), driver mutations in subgroups of tumors (Castro-Vega et al., 2015), markers to track the relapse of treated patients (Madhavan et al., 2013), and metastasis (Kong et al., 2020), database generation in cell lines (Berg et al., 2017) as well as for use in precision medicine (Xie et al., 2018).
It will be of interest to also incorporate information on sequence variants in the miRNA and its interacting partners as a part of the global interactome identification, thereby serving the needs of precision and personalized medicine in a clinical setting. Development of miRNA regulatory pathways in a pan-cancer model and domestic animals will help pinpoint common/synergistic regulatory pathways and generate novel therapies targeting/modulating miRNAs on different human and zoonotic diseases (Pasquini and Kunej, 2019; Zhang et al., 2016a).
Attempts are also being made to generate markers from pan-cancer datasets by integrated clustering methods and to compare their effectiveness as well as similarities between the various methods (Wei et al., 2020). It will become imperative to detect cancers early using markers from multiomics data or to determine novel markers or patient outcome, which could be pan-cancer in nature (Mitra et al., 2020). It will also be advantageous to incorporate live patient data in real time using automated methods for integration and analysis of multiomics data, all of which can be a big step in accessing real-time data and apply the knowledge in personalized medicine. Tools that perform end-to-end analysis in a single package format are also needed, and attempts are being made in that direction, utilizing multiomics for wider applications (Allendes Osorio et al., 2020).
Currently, advancements in multiomics focus on the integration of data from different omics and novel algorithms/software are being developed, despite the various issues (data handling, annotation, design, assumptions, and statistical analysis) in data integration (Misra et al., 2019). Integration tools of multiomics operating on different principles (Sathyanarayanan et al., 2019) as well as the tools for data visualization and data repositories are under development (Subramanian et al., 2020), and several of these tools incorporate miRNA expression data in the integration of omics data. Some of the recently published tools, especially with respect to miRNA and/or proteomics, with their advantages, input omics datasets, and final output have been summarized in Table 6.
Contemporary Tools for MultiOmics Integration Including Either Proteomics or miRNA Sequencing Data
MiRNA interactome in real-time studies can provide a snapshot of the significant pathways being altered during initiation of tumorigenesis, progression to metastasis, making it a strong tool for personalized medicine. It could also be used in single-cell omics to characterize individual cells in that heterogeneous bulk of the tumor, equipping us with the knowledge to target resistant cells in that bulk tumor. With a growing interest in single-cell multiomics and its applications, several reviews have covered the developments (Ma et al., 2020; Chappell et al., 2018; Hu et al., 2018), but lack any specific methodology to cover post-transcriptional regulation at the single-cell level.
Conclusions
We have come a long way from predicting one target for one miRNA and experimentally verifying those using reporter assays to large-scale studies that identify bulk targets and their other regulating partners. There is ongoing research to overcome the difficulties in data integration with upcoming better tools for data integration and interpretation. This evergrowing list of tools indicates the dominating role of miRNAs in physiology, and patholog and the necessity to understand the full picture is growing even more as personalized therapy becomes the norm (Detassis et al., 2017).
This can be accomplished only by multiomics encompassing proteomics. MiRNAs from the huge network of interactome can act as biomarkers, therapy resistance, and prognostic markers, as well as therapeutic targets, and work well with conventional therapy. The reducing costs of omics technologies can go all the way in making the miRNAs a deciding factor for personalized and cancer therapy.
Footnotes
Acknowledgment
We thank for the research fellowships from the Ministry of Human Resource Development, Government of India, to Ms. K J Sindhu, and Indian Council of Medical Research to Dr. Venkatesan Nalini (project number BMS/4566/2019).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Funding Information
No funding was received for this work.