
Abstract
The human hypothalamus is central to the regulation of neuroendocrine and neurovegetative systems, as well as modulation of chronobiology and behavioral aspects in human health and disease. Surprisingly, a deep proteomic analysis of the normal human hypothalamic proteome has been missing for such an important organ so far. In this study, we delineated the human hypothalamus proteome using a high-resolution mass spectrometry approach which resulted in the identification of 5349 proteins, while a multiple post-translational modification (PTM) search identified 191 additional proteins, which were missed in the first search. A proteogenomic analysis resulted in the discovery of multiple novel protein-coding regions as we identified proteins from noncoding regions (pseudogenes) and proteins translated from short open reading frames that can be missed using the traditional pipeline of prediction of protein-coding genes as a part of genome annotation. We also identified several PTMs of hypothalamic proteins that may be required for normal hypothalamic functions. Moreover, we observed an enrichment of proteins pertaining to autophagy and adult neurogenesis in the proteome data. We believe that the hypothalamic proteome reported herein would help to decipher the molecular basis for the diverse range of physiological functions attributed to it, as well as its role in neurological and psychiatric diseases. Extensive proteomic profiling of the hypothalamic nuclei would further elaborate on the role and functional characterization of several hypothalamus-specific proteins and pathways to inform future research and clinical discoveries in biological psychiatry, neurology, and system biology.
Introduction
The human hypothalamus plays an essential role in human physiology, its regulation, not to mention in a host of common complex human diseases. A deep proteomic analysis of the normal human hypothalamic proteome, surprisingly, has been missing for such an important organ so far. This knowledge gap is important to address for a comprehensive understanding of human health and disease, not to mention toward discovery of novel diagnostics and therapeutics.
As a way of introduction of hypothalamus to the readers, it is anatomically located in the third ventricle, obliquely front and under the thalamus, with a stalk extending and connected to the pituitary gland (Lemaire et al., 2013). The hypothalamus comprises both gray and white matter, entailing groups of dense collections of cellular bodies (hypothalamic nuclei) and myelinated nerve fibres, respectively. The neuronal, as well as neurohumoral, connections between the hypothalamus and the other regions of the brain account for the integration of neuroendocrine, autonomic, and behavioral functions (Toni et al., 2004).
The most notable functions of the hypothalamus include maintenance of energy homeostasis, control of appetite (Ahima and Antwi, 2008; Timper and Bruning, 2017), water intake (Knepper et al., 2015), sleep–wake cycle (Van Drunen and Eckel-Mahan, 2021), thermoregulation (Zhao et al., 2017), and regulation of the hypothalamic–pituitary axis (Koizumi, 1996).
The hub of neuronal, as well as non-neuronal, networks originating from the hypothalamus includes several synaptic junctions, which are essentially plastic in nature to accommodate the diverse homeostatic variations of the body (Bains et al., 2015; Dietrich and Horvath, 2013; Levy and Tasker, 2012). This brings to question the contribution of the hypothalamus toward the metaplasticity of the human brain, which has so far been attributed only to the cerebral cortex, hippocampus, and olfactory bulb (Bocci et al., 2014; Jones et al., 2016; Lledo et al., 2006; Muller-Dahlhaus and Ziemann, 2015; Ni et al., 2014).
The intriguing and relatively novel phenomenon of adult neurogenesis in the mammalian hypothalamus, and the role of synaptic plasticity in maintaining its regular functions, warrants the need to explore the molecular dynamics of the human hypothalamus proteome (Migaud et al., 2015; Rojczyk-Golebiewska et al., 2014; Sousa-Ferreira et al., 2014).
To understand the role of hypothalamus in mental disease-related brain pathology, meta-analyses have been conducted revealing alterations in the gross anatomy of the human hypothalamus (Bernstein et al., 2019). For instance, a reduction in the volume of the mammillary body of the human hypothalamus was observed in major depressive disorder (Bernstein et al., 2012).
In addition, variations in the expression of hypothalamic neuropeptides such as oxytocin (OXT) and vasopressin (AVP) were reported in psychiatric disorders such as schizophrenia (LaCrosse and Olive, 2013). Perturbations in energy homeostasis have also been linked to hypothalamic defects in neurodegenerative disorders such as Alzheimer's disease (AD), Huntington's disease, amylotropic lateral sclerosis (ALS), and dementia (Vercruysse et al., 2018). A dysfunction in the orchestration of hypothalamic signals in regulation of food intake and energy expenditure has been consistently described in these neurological disorders.
In addition, a reduction in the orexinergic neurons has also been linked to the sleep deprivation associated with AD patients (Fronczek et al., 2012). An atrophy of anterior and posterior hypothalamus and presence of TDP-43 or Tau protein aggregates in lateral hypothalamus correlated with a reduced body mass index in ALS patients (Cykowski et al., 2014; Gorges et al., 2017). Thus, the understanding of the human hypothalamus at the proteome level will greatly enable the elucidation of disease pathology in the fields of biological psychiatry and clinical neurology.
Owing to the developments in the field of science, emerging high-throughput OMICs platforms have been extensively used in the past two decades for the molecular characterization of the mammalian hypothalamus (Chin, 2007; Colgrave et al., 2011; Iqbal et al., 2013; St-Amand et al., 2011; Stelzhammer et al., 2012; Zhang et al., 2019). In addition, the proteomic profiles of certain hypothalamic nuclear regions from rat or mouse brains have been performed to focus on certain physiological responses such as obesity, sleep deprivation, and disturbance in the body homeostasis (Bora et al., 2008; Chiang et al., 2014; Hatcher et al., 2008). Despite these efforts, the elucidation of the human hypothalamus proteome as a whole is lacking in the literature. Availability of such data would act as a resource to understand the diverse range of functions attributed to the hypothalamic region.
In this study, we report on the global proteomic profile, including multiple post-translational modifications (multiPTMs), and a proteogenomic profile of the hypothalamus region from the adult human brain using a high-resolution mass spectrometry approach. In addition to identifying a large number of proteins expressed in the hypothalamus, we also provided peptide evidence for novel protein-coding regions of the genome, which was used to revise the current protein sequences and identify novel proteins and proteins coded by pseudogenes.
Materials and Methods
Sample collection
All samples were obtained from the Human Brain Tissue Repository (HBTR), National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore. The hypothalamus tissue was dissected from three individuals with histopathologically normal adult brain samples as determined by trained qualified neuropathologists at HBTR (Supplementary Table S1). The dissected tissue sections were stored at −80°C until further analysis. The study was conducted with the approval of the Ethics Committee at NIMHANS, Bangalore. Written consents were obtained from the close relatives of the deceased tissue donors.
Protein extraction
The frozen tissue samples were lysed in tissue lysis buffer (TLB: 4% sodium dodecyl sulfate [SDS]) by sonication using a probe sonicator. The sonicated tissue suspended in TLB was then subjected to heating at 95°C for 10 min. This was followed by centrifugation at 12,000 rpm for 10 min, after which the clear supernatant containing the proteins was collected in fresh vials. The protein estimation was carried out using the Bicinchoninic Acid (BCA) Kit (Thermo Scientific Pierce). Equal amounts of protein were pooled from the three donor tissues and were used for further analysis. These strategies were similar to the protocol followed by the analysis of a draft map of the human proteome (Kim et al., 2014).
Protein and peptide level fractionation
The protein level fractionation was performed using the in-gel digestion technique as described in Mohanty et al. (2020). Briefly, around 100 μg of pooled lysate was resolved on 10% SDS-polyacrylamide gel electrophoresis (PAGE) gel, and 24 gel bands/fractions were excised. The bands were cut into 1 mm pieces and suspended in destaining solution (40 mM ammonium bicarbonate in 40% acetonitrile [ACN]). The completely destained bands were then subjected to reduction using 5 mM dithiothreitol (DTT; in 50 mM tetraethylammonium bicarbonate [TEABC]) at 60°C for 30 min followed by alkylation in the dark for 20 min in 20 mM iodoacetamide (IAA; in 50 mM TEABC).
The protein digestion was carried out overnight at 37°C using Sequencing grade modified trypsin (Promega, Madison, WI, USA). The peptides were then extracted using 40% ACN and desalted using C18StageTips (3M Empore high-performance disks) (Rappsilber et al., 2003). The desalted peptides were then vacuum dried and stored at −80°C until mass spectrometry runs were performed.
The peptide level fractionation was carried out with basic-reversed phase liquid chromatography. A total of 300 μg of pooled lysates were subjected to reduction and alkylation, as mentioned above. Protein precipitation was carried out with chilled acetone (1:7). The protein pellet was resuspended in 50 mM TEABC and was subjected to overnight trypsin digestion using sequencing grade modified trypsin (1:20 v/v; Promega). The digested peptides were lyophilized before the basic pH reversed-phase liquid chromatography (bRPLC)-based fractionation.
The bRPLC solvent A (10 mM TEABC, pH 8.5) was used to reconstitute the dried peptides, which were then loaded on a Waters XBridge C18 column (5 μm, 250 × 4.6 mm; Waters Corporation, Milford, MA, USA) using the Agilent 1200 binary pump (Agilent Technologies, Santa Clara, CA, USA). The peptides were separated on 120 min long gradient of 5–100% bRPLC solvent B (10 mM TEABC in 95% ACN, pH 8.5) with a flow rate of 0.5 mL/min. A total of 96 fractions were collected, which were concatenated to a final of 12 fractions. The peptide fractions were vacuum dried and stored at −80°C until further analysis.
Tandem mass spectrometry analysis
The digested peptide fractions (n = 36) were analyzed using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, Bremen, Germany) coupled with a Thermo Scientific Easy-nLC 1200 (Thermo Scientific). Each of the peptide fractions was resuspended in 0.1% formic acid (Solvent A), loaded on a C18 packed nanoViper trap column (100 Å, 3 μm, 75 μm × 2 cm; Thermo Scientific), and the flow rate was fixed at 4 μL/min of solvent A. The peptides were resolved on a C18 packed Easy-Spray RLC analytical column (2 μm, 15 × 50 μm; Thermo Scientific) with 120 min long gradient of 5–35% of solvent B (0.1% formic acid in 95% ACN) and flow rate of 300 nL/min.
The data-dependent acquisition mode was used in the positive ion mode to acquire data. The scan range was set at 400–1600 m/z, and a resolution of 120,000 was used on the Orbitrap mass analyzer for MS1 runs. The ion injection time of 55 msec, dynamic exclusion of 40 sec, and automated gain control (AGC) target of 400,000 were used at MS1. The top 10 intense precursor ions were selected for tandem mass spectrometry (MS/MS) analysis and subjected to higher energy collisional dissociation with a normalized collision energy of 33%. The fragment ion detection was also carried out in the Orbitrap mass analyzer with a resolution of 60,000, ion injection time of 75 msec, and isolation window of 1.6.
Database searches and data processing
The liquid chromatography-based tandem mass spectrometry (LC-MS/MS) data for all 36 fractions (24 of in-gel and 12 of bRPLC) were carried out in the Proteome Discoverer 2.1 software (Thermo Scientific) against the Human RefSeq 81 protein database (with 110,502 protein sequences inclusive of 116 common contaminants arising from standard sample processing steps and exposure to dust, skin, or hair). Both SequestHT (v 1.0) and Mascot (v 2.5.1; Matrix Science, London, United Kingdom) search algorithms were used. The search parameters were set with trypsin as the proteolytic enzyme, two allowed missed cleavages, and a minimum seven amino acid length of a peptide.
The dynamic modifications were set as protein N-terminal acetylation and oxidation of methionine, and carbamidomethylation of cysteine was set as the static modification. The precursor mass tolerance was set at 10 ppm, and the fragment mass tolerance was set to 0.05 Da. The cutoff false discovery rate (FDR) was set to 1% at the protein, peptide, and peptide spectral match (PSM) levels when the data were searched against the decoy database.
Bioinformatics analysis
Proteins identified were subjected to Gene Ontology-based functional classification to know the enrichment of biological processes, molecular functions, and cellular components using clusterProfiler tool (Yu et al., 2012). The relative abundances of identified proteins were determined using an intensity-based absolute quantitation (iBAQ) (Schwanhausser et al., 2011). The intensities of the observed tryptic peptides of a given protein were summed and then divided by the number of theoretically observable tryptic peptides using an in-house python script. SignalP 5.0 (http://www.cbs.dtu.dk/services/SignalP/) was used to predict the presence of signal peptides among the identified proteins.
MultiPTMs of hypothalamus proteins
We carried out a global multiPTM search to identify the commonly occurring post-translational protein modifications in the human hypothalamus. The strategies were adopted from previous such investigations from our group using publicly available proteomic data of AD, glioma, and oral cancer (Deolankar et al., 2021; Palollathil et al., 2021; Rex et al., 2021).
Although we have not used any enrichment protocol, there will be several mass spectra for peptides with different types of post-translational modifications (PTMs), which can be captured through a multiPTM search. Compared to the proteomic search, a multiPTM search was more demanding in terms of search time and computational features. The unassigned spectra obtained after a proteomic search were used for a subsequent search with additional parameters of multiPTMs, which included the addition of the ptmRS node in the processing workflow on Proteome Discoverer 2.1 and the dynamic modifications such as (1) S, T, Y-phosphorylation; (2) K-acetylation; (3) K, R-methylation; (4) N, Q-deamidation; and (5) R-citrullination. The modified PSMs with a ptmRS score >75% were filtered and input into the PTM-Pro tool for the prediction of high confidence PTMs (Patil et al., 2018).
Proteogenomic analysis
The unassigned spectra from the proteomic and multiPTM searches were further used for a proteogenomic analysis. A custom reference database was generated, using a three-frame translation of the GENCODE genome annotation (v26), inclusive of transcripts, pseudogenes, noncoding RNAs, and N-terminal peptides. This database included sequences of minimum length ≥7 amino acids, translated from stop-to-stop codons. The other search parameters were kept similar to the previous proteomic search. The resultant peptides from this search were filtered out if they were mapped to any known protein or had an isobaric mass ambiguity. The remaining peptides that were uniquely identified through the custom database were taken forward for analysis. These peptides were termed as Genome Search Specific Peptides (GSSPs) and were selected only if they met a 1% FDR threshold and had a minimum of two PSMs.
The spectra quality of each GSSP was verified adhering to the manual assessment criteria. Briefly, for a peptide to be considered to have agreeable spectral quality it must have a higher number of PSMs, preferable length of 7–25 amino acids, assignment of continuous b-y ions, an Xcorr ≥3 and Ion score ≥40, and lower signal to noise ratio (Antil et al., 2021; Elias et al., 2005).
The Integrative Genome Viewer (IGV) v2.3 was used for visualizing the reference genome, known genes, and the GSSPs for conducting the analysis. Based on the genomic coordinates and BLASTp (Basic Local Alignment Search Tool for proteins) alignment analysis, we categorized the GSSPs into: (1) overlapping with already annotated genes, (2) protein extensions (C-terminal or N-terminal), (3) alternate frame of translation, (4) novel protein-coding isoform/exon/gene, (5) short open reading frames (sORFs), and (6) evidence for pseudogene with protein-coding potential. We report the most consensus sequence and related genomic coordinates for the protein revisions, based on transcripts visualized on IGV and the homologous alignment observed on BLASTp analysis.
Data availability
The raw data acquired during this study were deposited on the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) and can be accessed with the PRIDE dataset identifier PXD026879.
Results and Discussion
The human hypothalamus proteome
We carried out a global proteomic analysis of the adult human hypothalamus using in-gel digestion and bRPLC-based fractionation strategies, followed by the data acquisition on an Orbitrap Fusion Tribrid high-resolution mass spectrometer (Fig. 1). This led to the identification of 5349 proteins (Supplementary Table S2). We also confirmed the translational start sites of 902 proteins based on the N-terminal acetylated peptides identified in the study.
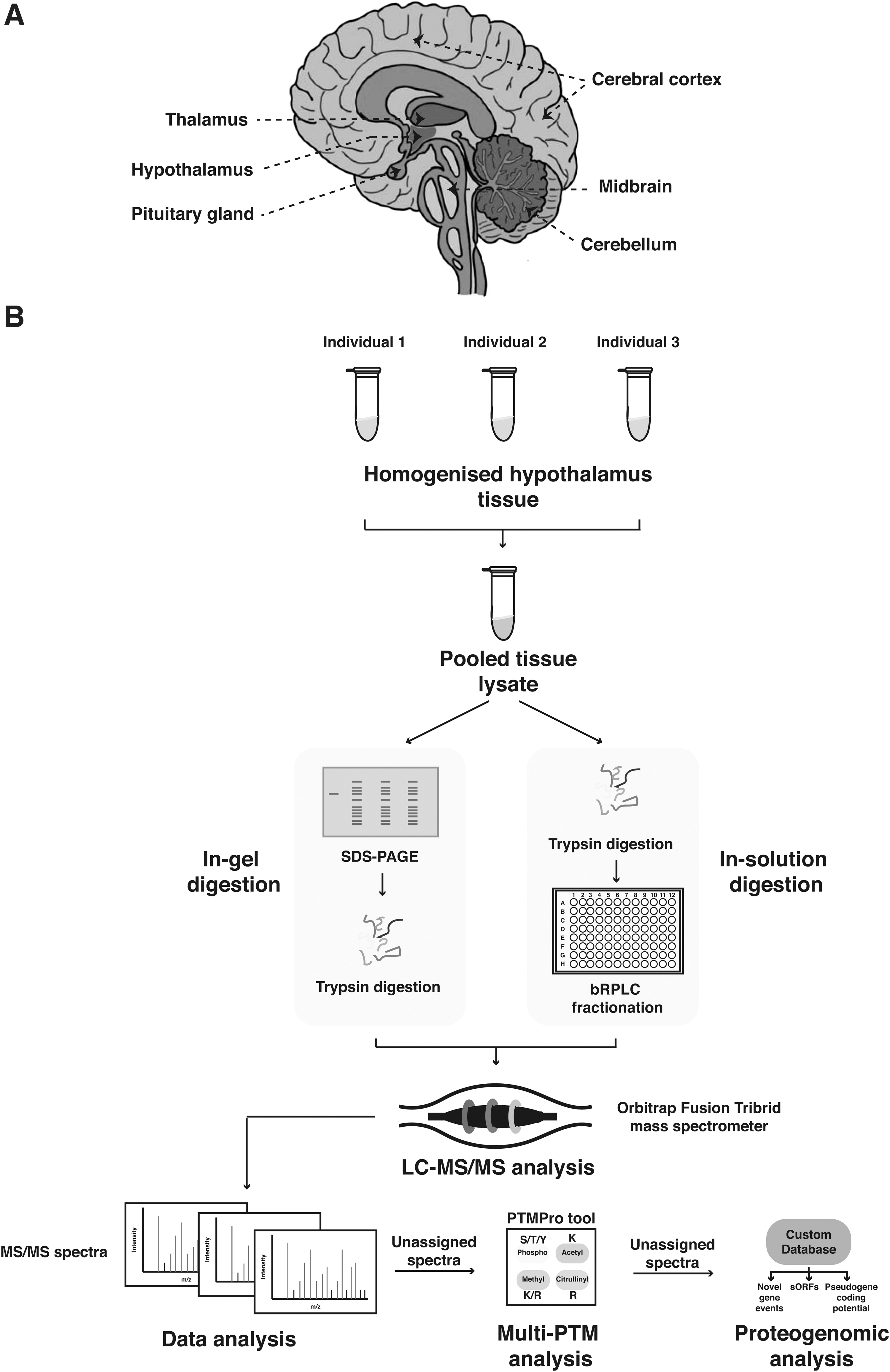
Schematic representation of the human brain (medial view) and the overall proteomics workflow used in the study.
We next performed the SignalP (v 5.0) analysis to identify the secretory proteome of the human hypothalamus (Almagro Armenteros et al., 2019). We identified 628 proteins with signal peptides, which included several known neuropeptide hormones, synaptic transmission proteins, as well as neurotransmitter receptors. We also identified 785 mitochondrial proteins compared with the Human Mitocarta 2.0 database (n = 1158) (Calvo et al., 2016). Apart from this, 778 hypothalamus proteins were found to be known synaptic proteins as curated in the SynGO database (Koopmans et al., 2019) (Supplementary Fig. S1).
We subjected the hypothalamus proteins to label-free quantitation to estimate the relative abundances of these identified proteins. The complete list of proteins and the corresponding iBAQ values are provided in Supplementary Table S3. The myelin proteolipid protein (PLP1), which is also known to be specific to brain tissue, was found to be the highest abundant protein. Myelin basic protein (MBP), 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP), dihydropyrimidinase like 2 (DPYSL2), glial fibrillary acidic protein (GFAP), and ubiquitin C-terminal hydrolase L1 (UCHL1) were found to be other high abundant proteins.
In addition, we observed that the top 10% abundant proteins were associated with the cytoskeletal organization, regulation of cellular metabolic processes, myelination, and mitochondrial function. These include tubulin (TUBB), TUBA3D, TUBB2A, actin (ACTB), enolase-1 (ENO1), enolase-2 (ENO2), lactate dehydrogenase B (LDHB), ATP synthase F1 subunit beta (ATP5B), phosphoglycerate mutase 1 (PGAM1), malate dehydrogenase (MDH2), and so on.
Integration of human hypothalamus proteome with publically available omics data
There are several studies available on the hypothalamic proteome of rat, mouse, and bovine brains; however, a landscape of the human hypothalamus proteome is still uncharted. We compared our data with the rat hypothalamus proteome datasets (Iqbal et al., 2013; Pedroso et al., 2012; Stelzhammer et al., 2012). We observed that 77 and 594 proteins were common to Pedroso et al. (2012) and Stelzhammer et al. (2012), respectively, compared to human hypothalamus proteome data (Fig. 2A, B). These proteins include several enzymes related to metabolic processes and 65 Ras-related proteins.
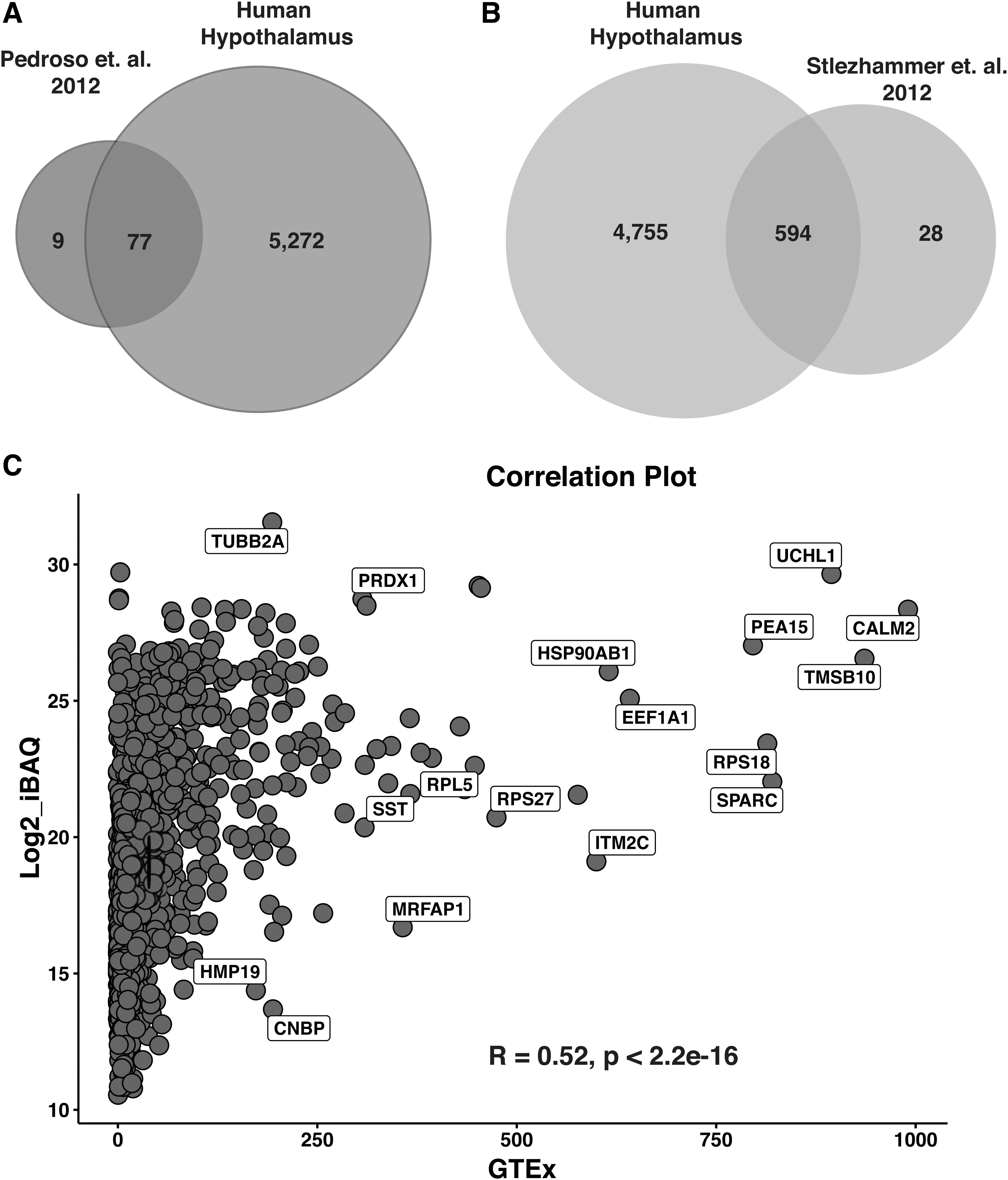
Comparison with previously published omics data.
Apart from these, several studies focussing on the neuropeptidomics of the rat hypothalamic nuclei such as ventromedial (Mo et al., 2006), supraoptic (Bora et al., 2008), and suprachiasmatic nuclei (SCN) (Hatcher et al., 2008) were also available (Supplementary Table S4). Similar studies on mouse SCN (Chiang et al., 2014) and bovine hypothalamus (Colgrave et al., 2011) have revealed the identification of 2112 proteins and 140 neuropeptides, respectively.
We next sought to compare and correlate the human hypothalamus proteomic data from this study with the human hypothalamus RNA-Seq data available on the GTEx database (https://gtexportal.org/home). Using Spearman's correlation method, we identified a rho value of 0.52 with a p-value <2.2e−16, between the iBAQ abundances of peptides identified in this study and the transcripts per million abundances from the GTEx dataset. This weak correlation between the transcriptome and proteome data can be attributed to the fact that the process of translation of proteins from the transcripts is regulated by several factors, such as splicing, post-transcriptional and PTMs, gene silencing, transcript turnover, and so on.
We observed the highest correlation for cytoskeletal organization proteins such as calmodulin 2 (CALM2) and thymosin beta 10 (TMSB10), as well as brain-specific protein UCHL1 (Fig. 2C). In addition, we also confirmed protein evidence for 73 of the top 100 hypothalamic genes listed in the GTEx database. Among the proteins reported to be elevated in the hypothalamus as per Human Proteome Atlas (HPA), we identified 20 proteins, inclusive of OXT, pro-melanin concentrating hormone (PMCH), secretagogin (SCGN), neuroglobin (NGB), phosphotriesterase-related (PTER), and neurotensin (NTS) (Supplementary Table S5).
PTMs of hypothalamic proteins
The unassigned spectra from the proteomic search were further utilized for detecting potential peptides with PTMs. The proteins represented by only these peptides might have been missed in the previous search. The incidence of PTMs such as citrullination, acetylation, phosphorylation, and methylation is essential for the normal development and functioning of the human brain. Variation in the PTMs or the expression of enzymes responsible for these modifications is often associated with psychiatric and neurodevelopmental disorders (Tapias and Wang, 2017).
The PTMPro analysis (v 2.0) led to the identification of 13,544 peptides corresponding to 2542 proteins with modifications of R/K-methylation, S/T/Y-phosphorylation, N/Q-deamidation, R-citrullination, and K-acetylation (Patil et al., 2018) (Fig. 3A and Supplementary Table S6). Overall, we observed a higher representation of R-citrullination, followed by S-phosphorylation and K-acetylation. The inclusion of these modifications in the search parameters aided in the identification of 191 additional proteins.
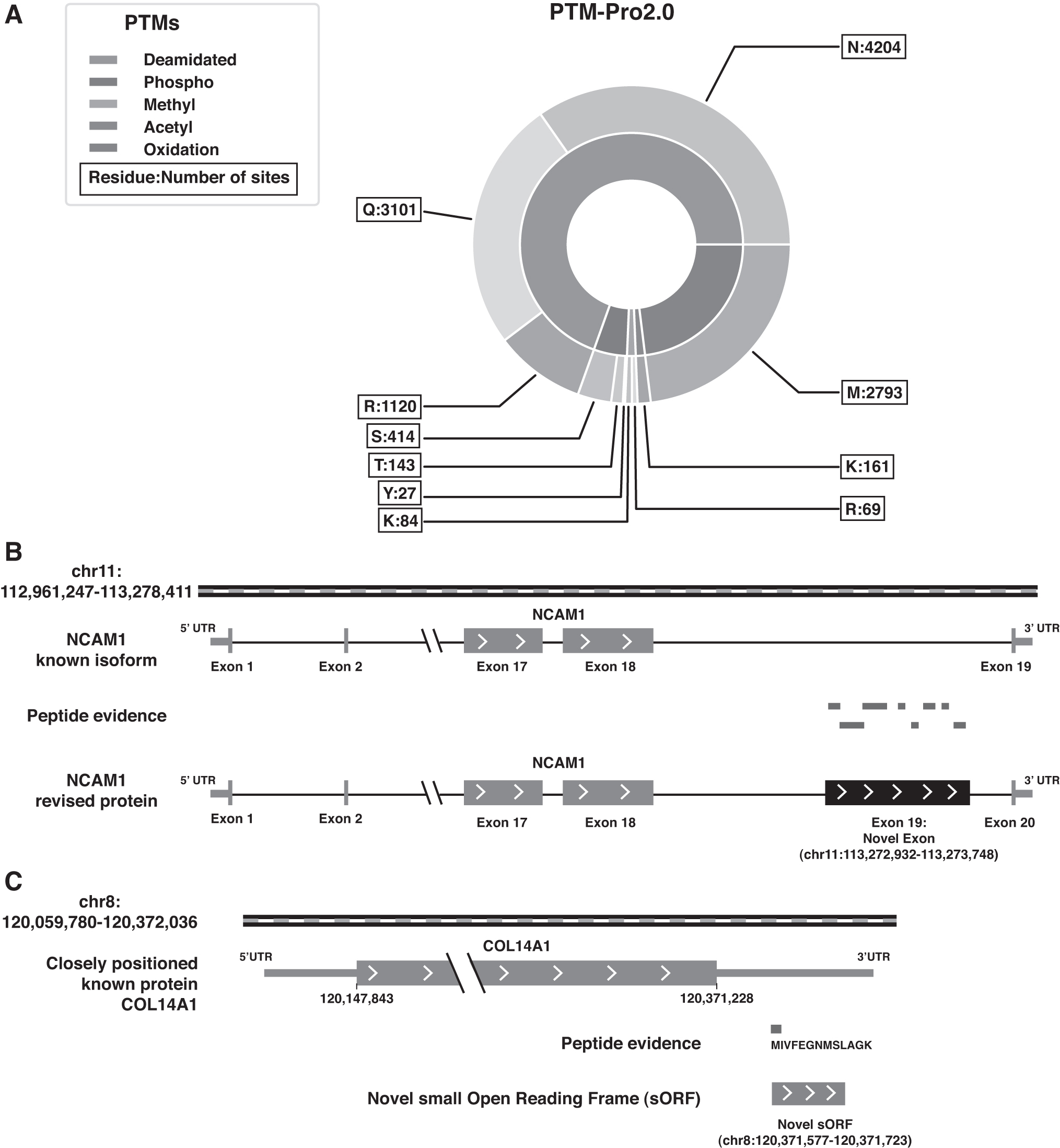
MultiPTM and proteogenomic analysis.
The brain has been shown to have the highest level of citrullination compared to other regions of the body (Lee et al., 2018; Nicholas et al., 2003; Pritzker et al., 1999). This can be attributed to the increased expression and activity of peptidylarginine deiminase enzyme, which is essential for conversion of arginyl to citrulline residues (Pritzker et al., 1999). Protein citrullination is known to be implicated in myelin formation and has a suggested role in the pathogenesis of neurodegenerative disorders such as multiple sclerosis and AD (Faigle et al., 2019).
Interestingly, citrullinated proteins were either found to be cytoskeletal proteins or related to the cellular structure (Conrad et al., 2010; Jiang et al., 2013; van Beers et al., 2013). We observed a similar trend in the human hypothalamus. Our analysis revealed the potential citrullinated sites on 510 proteins, among which 70 proteins were common to Lee et al. (2018), which has reported 274 citrullinated proteins of the human brain. Consistent with the literature, we observed the maximum number of citrullinated peptides on GFAP. We confirmed already known citrullinated sites on GFAP such as R36, R105, R173, R217, R258, R270, R276, R287, and R416 and identified other potential citrullination sites (Jin et al., 2013) (Supplementary Table S7). Among other proteins with a higher representation of citrullinated peptides were MBP, TUBB, CNP, and creatine kinase B (CKB).
Acetylation of lysine residues is a PTM known to be conserved across species (Kim et al., 2006). Investigation of the human acetylome by Choudhary et al. (2009) reported 1750 proteins with more than 3600 acetyl sites on lysine. We identified a total of 115 proteins in the human hypothalamus to have K-acetyl sites, among which 53 proteins were common to those identified in Choudhary et al. (2009). In line with previous studies, we observed several acetylated peptides to be on enzymes involved in metabolism and mitochondrial proteins, such as glutamate dehydrogenase-1 (GLUD1), aldolase fructose-bisphosphate A (ALDOA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ENO2, glutamic-oxaloacetic transaminase 2 (GOT2), MDH2, pyridoxal phosphatase (PDXP), pyruvate kinase M1/2 (PKM), and superoxide dismutase 2 (SOD2) (Baeza et al., 2016; Kim et al., 2006).
Apart from acetylation and citrullination, another interesting PTM was that of the R/K methylation. We identified 84 K-methyl and 69 R-methyl sites on 160 peptides corresponding to 105 proteins. One of the interesting findings was the identification of methylation sites on R, as well as K, on a single peptide in the case of proteins such as heat shock protein family A member 2 (HSPA2), cytochrome c1 (CYC1), PKM, ATPase H+ transporting V1 subunit A (ATP6V1A), and TUBB. Among others, we identified two mono-methyl sites at K79 and K84 in a single peptide corresponding to elongation factor 1-alpha 2 (EEF1A2) protein. These sites have been previously annotated as possible trimethyl and dimethyl sites, respectively (https://www.phosphosite.org/homeAction.action).
We also identified R-methyl sites on several neuronal proteins such as GFAP, synapsin I (SYN1), neurogranin (NRGN), CNP, L1 cell adhesion molecule (L1CAM), neural cell adhesion molecule 2 (NCAM2), and neuronal growth regulator 1 (NEGR1). In addition, several cytoskeletal and heat shock proteins were found to be enriched in R-methyl sites. Some of the notable examples being heat shock protein family A member 8 (HSPA8) and MBP, where the former is represented by the highest number of PSMs and the latter is represented by the highest number of R-methyl sites identified per protein (n = 4).
We identified a total of 584 unique phospho site modifications, with 414 S-phospho, 143 T-phospho, and 27 Y-phospho sites. We identified phosphorylation sites on proteins such as microtubule-associated protein 2 (MAP2), SYN1, neuromodulin (GAP43), and MBP, which have previously been reported as phosphoproteins in the human brain (Walaas et al., 1989). Several mitochondrial proteins such as pyruvate dehydrogenase E1 component subunit alpha (PDHA1), cAMP-dependent protein kinase catalytic subunit alpha (PRKACA), and cytochrome b-c1 complex subunit 1 (UQCRC1) were identified to have peptides with S, T, and Y phospho sites, respectively.
However, a serine protease, mitochondrial (HTRA2) was found to have phospho modifications on all three residues in a single peptide (VRLLsGDtyEAVVTAVDPVADIATLR). We also observed several cytoskeletal proteins showing both citrullination and phosphorylation in a single peptide. These include MBP and collagen alpha-1 (COL1A1). Apart from these, we also identified phosphosites in other neuronal proteins such as GFAP, synapsin 2 (SYN2), synaptic vesicle glycoprotein 2A (SV2A), synaptojanin 1 (SYNJ1), and CNP among others.
Proteogenomic analysis of the human hypothalamus
We performed the proteogenomic analysis on the unassigned MS/MS spectra from the human hypothalamus proteomics and multiPTM search data. We identified an additional 425 protein-coding genes compared to the proteins identified in the proteomics search. A total of 291 GSSPs were identified and further categorized into probable gene correction events. Among these, we confirmed 28 peptides to confer evidence for 10 predicted pseudogenes with coding potential, 4 novel protein-coding (3 novel isoforms and 1 novel sORF-encoded peptide [SEP]), and 3 protein correction events (1 novel exon, 1 alternate frame of translation, and 1 N-terminal protein extension).
Among the examples with multiple peptide evidence, we identified a novel exon in the NCAM1 supported by eight peptides (Fig. 3B) and an alternate frame of translation for immunoglobulin kappa constant (IGKC) supported by four peptides (Supplementary Table S8). In addition, we identified novel isoforms for synaptotagmin 8 (SYT8), diacylglycerol kinase kappa (DGKK), and endogenous retrovirus group K3 member 1 (ERVK3–1) proteins and an N-terminal extension of FKBP prolyl isomerase 8 (FKBP8). The parent genes for most of the pseudogenes with evidence for coding potential were observed to be enzymes and cytoskeletal proteins.
Another interesting example was that of a sORF with 48 amino acid residues, identified through the peptide “MIVFEGNMSLAGK” positioned at the 3′ untranslated region of the already known protein collagen type XIV alpha 1 chain (COL14A1) on chromosome 8 (Fig. 3C). According to the HPA, COL14A1 is most abundant in the pituitary gland, spinal cord, and the brain stem, among human brain regions. In addition, a study on mice lacking hypothalamic orexin has shown downregulation of COL14A1 to be related to sleep disorders such as narcolepsy (Honda et al., 2009).
There are several studies providing evidence for such sORFs to be a substantial and functional part of the eukaryotic genome (Frith et al., 2006; Mackowiak et al., 2015; Pueyo et al., 2016; Saghatelian and Couso, 2015). As shown by Oyama et al. (2007), a short human SEP identified through mass spectrometry (MS) analysis was 88 amino acids long. A previous study has reported that around 57% of the human SEPs possess non-AUG start sites (Slavoff et al., 2013). Manual examination of the coding sequence on the Integrative Genome Viewer (IGV) revealed the start site to be AUG for the SEP identified in this study. This necessitates the need to further investigate the existence of other brain-specific sORFs and their probable roles in the normal functioning or disease correlation of the human brain.
Functional characterization of the human hypothalamus proteome
To functionally characterize the hypothalamic proteins, we performed the gene set enrichment analysis using the ClusterProfiler (v 3.18.0) package in R (v 4.0). We observed a higher representation of proteins in biological processes such as protein targeting, macroautophagy, metabolic processes, and vesicle organization (Fig. 4A and Supplementary Table S9). Among these, the association of the hypothalamus and autophagy is one of the well-established phenomena. Studies in mice have shown that the deletion of autophagy-related (ATG) essential genes in the hypothalamus leads to obesity and energy metabolism disorders (Kaushik et al., 2011).
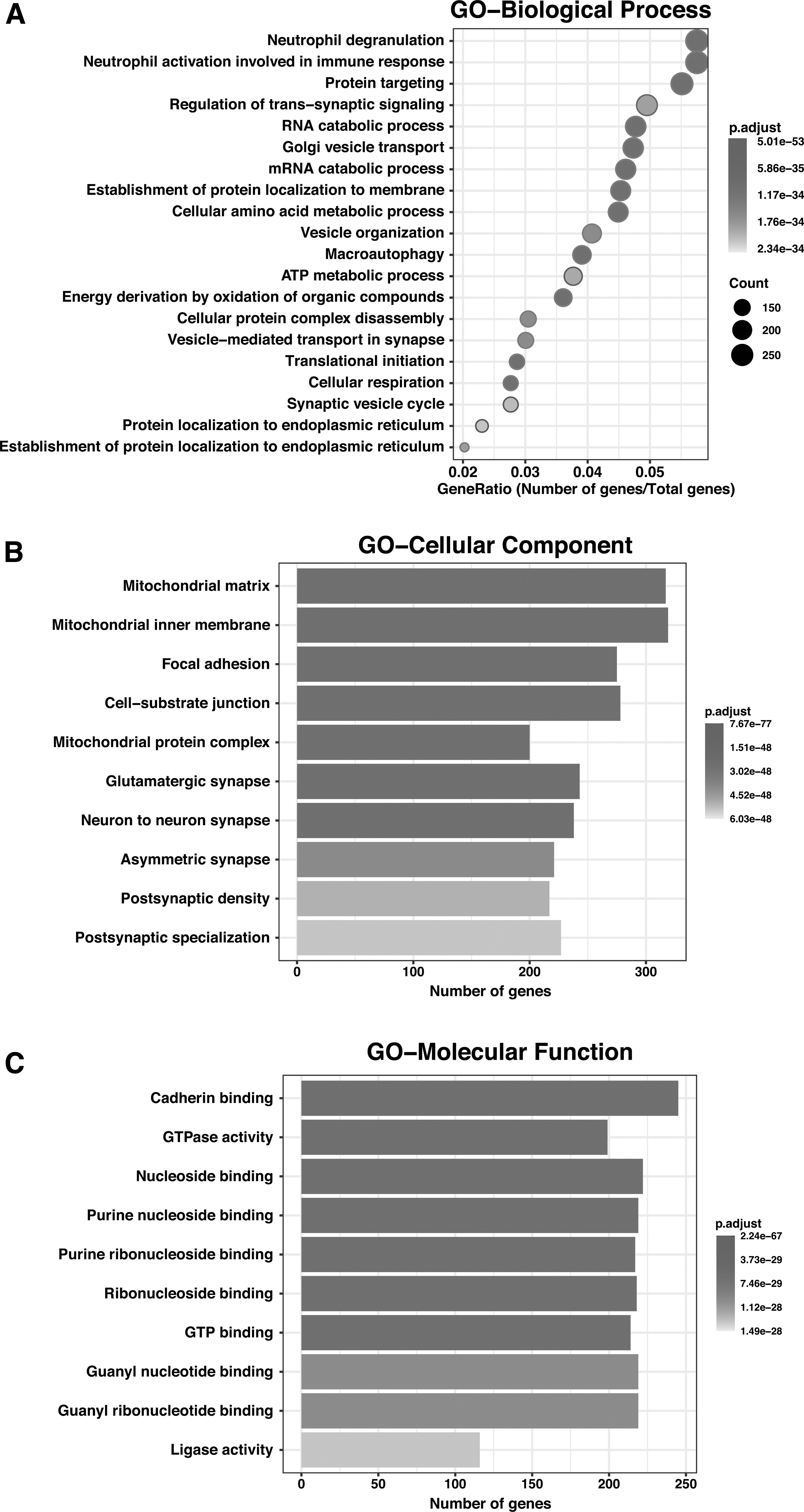
Gene Ontology analysis.
The most enriched molecular functions included cadherin binding, GTPase activity, nucleoside binding, ligase activity, and phospholipid binding to name a few (Supplementary Table S10). Among the cellular components, the maximum represented were the mitochondrial matrix and membrane, as well as synapse and postsynaptic density (Fig. 4B, C and Supplementary Table S11). This corroborates with our previous analysis (compared with Mitocarta and SynGO databases), which revealed a high representation of mitochondrial and synaptic proteins in comparison with the respective databases.
Human hypothalamic pathways enriched in the proteomic data
The pathway enrichment analysis performed using ClusterProfiler with KEGG as background database revealed several significantly enriched pathways (Yu et al., 2012). A list of the statistically significant enriched pathways from the human hypothalamus dataset is provided in Supplementary Table S12. Interestingly, much like the different hypothalamic functions of energy balance, fat metabolism, feeding behavior, obesity, autophagy, neuroprotection, and maintenance of reproductive physiology, the enriched pathways are interlinked to one another. A few of the most relevant physiological phenomena are discussed in the upcoming sections with reference to their corresponding enriched pathways from this analysis.
Neuroendocrine function
Hypothalamus is known to be the control hub of the neuroendocrine system of the human body. The hypothalamic projection pathways into the hypothalamo–pituitary axis are very essential for several neuroendocrine functions regulated by the hypothalamus (Smith and Vale, 2006). The majorly studied neuroendocrine regulation with respect to the hypothalamo–pituitary axis includes the thyroid, adrenal, and reproductive systems (Koizumi, 1996). The regulation of homeostasis, energy balance, metabolism, and feeding control are important functions performed by the neuropeptide hormones synthesized and released by the hypothalamus (Roh et al., 2016). We identified neuropeptide hormones such as OXT, AVP, POMC, corticotropin-releasing hormone (CRH), somatostatin (SST), NPY, and galanin (GAL), to name a few (Fig. 5A).

Network and pathway enrichment analysis.
The pathway analysis revealed the enrichment of the oxytocin signaling pathway, as well as vasopressin regulated water reabsorption pathway, which is among the most widely studied neuropeptide pathways associated with the hypothalamus (Boone and Deen, 2008; Chatterjee et al., 2016). The role of both OXT and AVP in the modulation of the cortical circuits pertaining to social cognition, fear response, anxiety, and depression-like behavior has been demonstrated (Kirsch et al., 2005; Neumann and Landgraf, 2012; Zink et al., 2010). Thus, the synergistic actions of these hypothalamic neuropeptide pathways might be essential for the regulation of social behavior.
Interestingly, we identified the apelin signaling pathway to also be enriched in this dataset. Studies in rats have shown the counteractive effect of apelin signaling to the vasopressin signaling mediated water reabsorption pathway (Hus-Citharel et al., 2014). Thus, the identification of both these pathways in the human hypothalamus proteome suggests collective activity in the regulation of water intake and reabsorption by the hypothalamus. In addition, apelin signaling is known to increase the secretion of both AVP and CRH, which essentially regulate the hypothalamic-pituitary-adrenal axis in modulating stress (Dagamajalu et al., 2021; Newson et al., 2009; Taheri et al., 2002).
Apart from the neuropeptide hormones, we also identified a hub of peptide hormone processing proteins (Fig. 5A). Several proteins associated with neurotransmitter biosynthesis, neuron projection, folate biosynthesis, and amine-derived hormones were also identified to be a part of the human hypothalamus proteome. This is supported by the enrichment of pathways such as protein processing in the endoplasmic reticulum, biosynthesis of amino acids, axon guidance, synaptic vesicle cycle, protein export, and regulation of actin cytoskeleton (Supplementary Table S12). Furthermore, we also observed enrichment of dopaminergic synapse, growth hormone secretion, synthesis, and action, GnRH signaling pathway, and thyroid hormone signaling.
Energy metabolism
Another important function of the hypothalamus is its role in the regulation of feeding behavior. The neuropeptides NPY and POMC along with AGRP and CART (CARTPT), also identified in this study, have well-established roles in the regulation of appetite, satiety, and energy metabolism. We observed an enrichment of AMPK signaling and PPAR signaling pathways, both of which also are known regulators of food intake and energy expenditure (Andersson et al., 2004; Ryan et al., 2011; Sarruf et al., 2009). AMPK is also known to act as a nutrient/glucose sensor in the hypothalamus (Claret et al., 2007; Lockie et al., 2018; Minokoshi et al., 2004). Thus, AMPK aids in the regulation of glucose metabolism, including glucose uptake for gluconeogenesis and stimulation of glycolysis, which are among the enriched pathways identified in this study.
We observed enrichment of HIF1 signaling, insulin signaling, thyroid hormone signaling, and thermogenesis pathways in the human hypothalamus proteome. The HIF1 signaling is associated with glucose-sensing/uptake in the hypothalamus (Varela et al., 2017; Zhang et al., 2011), as well as regulation of energy expenditure and hence obesity (Gaspar and Velloso, 2018; Gaspar et al., 2018). We identified the hypoxia-inducible factor 1 subunit alpha inhibitor (HIF1AN) protein, which is an important regulator of the HIF1 activity. It is known to be connected to metabolic regulation (Zhang et al., 2010).
However, the exact mechanism of its action is still unclear. The insulin receptors in the hypothalamus are also known to have a role in stimulating the POMC neurons toward inhibition of food intake (Belgardt et al., 2008). Thyroid hormone signaling and thermogenesis also have links to the hypothalamic AMPK signaling (Contreras et al., 2016; Lopez et al., 2010). Thus, the enrichment of these pathways in the hypothalamus points toward the synergistic actions of all these pathways in controlling satiety, hunger, and energy metabolism of the body.
Macroautophagy
Macroautophagy is an important cellular function that is essential for the maintenance of intracellular homeostasis and for keeping stress-related metabolic disorders at bay (Mehrpour et al., 2010). Thus, macroautophagy or more commonly referred to as autophagy has a key role in the normal functioning of the hypothalamus. We identified several ATG pathways to be enriched in the human hypothalamus, including the phagosome, lysosome, phagocytosis, ubiquitin-mediated proteolysis, proteasome, and protein processing in the endoplasmic reticulum (Supplementary Table S12).
In this study, we identified mediators of both classical and alternative pathways for autophagy in the human hypothalamus. The classical pathway is observed in the case of constitutive, as well as starvation and feeding behavior-related autophagy (Komatsu et al., 2005); however, the alternative pathway is mainly stress induced (Nishida et al., 2009). We identified several evolutionarily conserved ATG proteins, including ATG3, ATG4B, ATG5, ATG7, ATG9A, ATG12, ATG13, ATG16L1, and ATG101 (Fig. 5B).
We also identified other proteins involved in all the stages of autophagy, such as initiation/nucleation, elongation, and autophagosome maturation. We identified the kinase complex components required for the recruitment of the lipids to the isolation membrane in the nucleation phase of autophagosome formation. These include Beclin-1 (BECN1), phosphatidylinositol 3-kinase catalytic subunit type 3 (Vps34/PIK3C3), phosphoinositide-3-kinase regulatory subunit 4 (Vps15/PIK3R4), and RAS like proto-oncogene B (RALB). Along with these, we identified components of the other major autophagosome initiation complex ULK1-ATG13-FIP200/RB1CC1 (Itakura and Mizushima, 2010).
Apart from these, we identified the ATG8 family LC3 proteins, microtubule-associated protein 1 light chain 3 alpha (MAP1LC3A) and beta (MAP1LC3B), known to localize on the autophagosome membranes after being conjugated to phosphatidylethanolamide (PE) through the action of ATG4B, ATG7, ATG3, and the ATG12-ATG7-ATG16L complex. This ubiquitin-like protein complex is associated with autophagosome elongation. In addition, we identified several players of selective autophagy, such as sequestosome 1 (SQSTM1), optineurin (OPTN), and Tax1 binding protein 1 (TAX1BP1), which help in identifying and sequestering ubiquitinated proteins into the autophagosome.
The regulation of autophagy in the hypothalamus has been frequently linked to feeding behavior, energy expenditure, and body weight. Studies in mice have shown the effects of the presence of ATG7, one of the key players in autophagy initiation in AgRP and POMC neurons, on fat accumulation and obesity (Kaushik et al., 2011; Quan et al., 2012). We also identified AMPK (PRKAA1) protein, TSC complex subunit 2 (LAM/TSC2), and regulatory associated protein of MTOR complex 1 (RPTOR), which are known to be associated with alternate pathways regulating autophagy based on the cellular energy status (Meley et al., 2006; Oh et al., 2016).
Autophagy is also known to have a role in mechanistically mediating adult neurogenesis induced by physical exercise (Lee et al., 2012; Xi et al., 2016). The autophagy regulators such as BECN1 and ATG5, identified in this study, have also been established to be required for adult neurogenesis (Dhaliwal et al., 2017; Yazdankhah et al., 2014). The identification of autophagy, as well as neurogenesis-related pathways (as discussed later), in this study warrants the need to investigate the correlation between these two major functions of the hypothalamus.
Adult neurogenesis
Recent studies have shown the possibility of a neurogenic niche in the hypothalamus region, other than the already established adult neurogenesis sites at the subgranular zone (of the dentate gyrus of the hippocampus) and the subventricular zone (Cheng, 2013; Yoo and Blackshaw, 2018). The experiments conducted on neural stem cells isolated from adult rat hypothalamus laid the foundation for hypothalamic neurogenesis in adult mammals (Evans et al., 2002; Xu et al., 2005).
Due to the limited number of studies on hypothalamic neural stem cells, there is a dearth of understanding of the hypothalamic neurogenic niche. This study identified both neuronal stem cell markers such as nestin (NES), vimentin (VIM), notch receptor 2 (NOTCH2), hes family bHLH transcription factor 1 (HES1), and CD63 and markers of differentiated neurons and astrocytes such as GFAP, internexin neuronal intermediate filament protein alpha (INA), MAP2tubulin beta 3 (TUBB3), neurofilament light chain (NEFL), neurofilament medium chain (NEFM), and neurofilament heavy chain (NEFH). This suggests the presence of a combination of new, as well as differentiated, neurons in the adult human hypothalamus, which complies with previously published studies (Bolborea and Dale, 2013; Rojczyk-Golebiewska et al., 2014; Wei et al., 2002).
The activation of the hypothalamic neurogenic niche in response to a high-fat diet is well established (Lee et al., 2012). The enrichment of thermogenesis, fatty acid metabolism, and degradation pathways point toward the potential role of proteins in regulating brain fat accumulation and hence neurogenesis. Studies in mice have elucidated an increase in the GnRH levels to be coincidental with an increase in neurogenesis, thus promoting antiaging activity (Maggi et al., 2014; Zhang et al., 2013). We also identified enrichment of longevity regulation pathway and GnRH signaling pathways in our data, which suggest the role of hypothalamic neurogenesis in neuroprotection and antiaging. Heat-induced neurogenesis in rats has been correlated with the expression of GABAergic and glutamatergic markers (Matsuzaki et al., 2017).
We have observed enrichment of GABAergic, as well as glutamatergic, synaptic pathways in the human hypothalamus proteome data suggesting an increased representation of these markers in the adult human hypothalamus.
The potential synthesis and release of several neuropeptides to postnatal neurogenesis in the hypothalamus have been elucidated in other mammals (Bakos et al., 2016; Iqbal et al., 1995; Rankin et al., 2003). The most studied among them being OXT and AVP. Among other neuropeptides identified were NPY, CARTPT, AGRP, and POMC, which have a role in the differentiation of hypothalamic progenitor cells (Sousa-Ferreira et al., 2011, 2014). Furthermore, we identified many neural growth factor receptors which have been known to strongly correlate with the localization of newborn neurons (Kokoeva et al., 2005; Pencea et al., 2001). These include ciliary neurotrophic factor receptor (CNTFR), epidermal growth factor receptor (EGFR), and fibroblast growth factor receptor 3 (FGFR3).
Several other studies conducted on pig hypothalamus and ring dove hypothalamus have introduced the implications of adult neurogenesis in reproductive physiology and courtship behavior, respectively (Chen and Cheng, 2007; Rankin et al., 2003). Apart from these, adult neurogenesis in the mammalian hypothalamus has also been explained as a consequence of brain injury, hormones such as estrogen and testosterone, voluntary exercise, and other environmental factors (Niwa et al., 2016; Pierce and Xu, 2010; Yulyaningsih et al., 2017). However, our understanding of the impact of adult neurogenesis on behavioral and other reproductive physiological aspects in humans is limited. Thus, further studies to understand the relevance of adult neurogenesis in the modification of hypothalamus circuitry for sensory information processing in the human brain is a rather exciting possibility.
Association of phospholipase D signaling in various aspects of hypothalamus functioning
One of the significantly enriched pathways is the phospholipase D (PLD) activity, known to be represented more in the hippocampus, hypothalamus, and cortical regions of the rat brain (Kobayashi et al., 1988). There are several elucidated roles of PLD activation in central nervous system physiology and related disease pathophysiology (Klein et al., 1995). The most interesting among these is its potential role in accelerating neurogenesis (Iannitelli et al., 2017). There are extensive discussions on the role of PLD1 in neural stem cell differentiation specifically in the neurite outgrowth (Kanaho et al., 2009; Park et al., 2015; Yoon et al., 2005).
In this study, we identified the downstream regulators of the MAP kinase pathway, which has been established as the major pathway inducing neurite outgrowth (Fukuda et al., 1995; Sarina et al., 2013). These include proteins such as microtubule-associated protein 1B (MAP1B) and MAP2, which essentially stimulate axonal outgrowth (Goold and Gordon-Weeks, 2005) and crosslinking of microtubules (Ray and Sturgill, 1987), respectively. This ultimately leads to the elongation of the axonal growth cone. The Rho-family GTPases identified in this study, such as ras homolog family member A (RHOA), cell division cycle 42 (CDC42), and Rac family small GTPase 1 (RAC1), have well-established roles in the regulation of PLD1 activity and by extension in neuronal differentiation in rats (Park et al., 2015).
We identified the downstream effectors of the PLCG1/PRKCA mediated PLD1 activation implicated in neural stem cell differentiation, including phospholipase A2 (PLA2G4C), cytochrome c oxidase subunit II (COX2), protein kinase A (PRKACA), prostaglandin E2 (PTGES2), and cAMP-responsive element binding protein 1 (CREB1). Among others, we even identified 3-phosphoinositide-dependent protein kinase 1 (PDPK1) and hippocalcin (HPCA), which are also known to aid in the calcium-dependent activation of PLD1 in neuronal differentiation of neural stem cells (Park et al., 2017).
Other roles of PLD include the regulation of hormone release from neuroendocrine cells (Vitale et al., 2001) and neurotransmitter release from neurons (Humeau et al., 2001). We identified the enrichment of synaptic vesicle cycle, cholinergic synapse pathways, the phosphatidylinositol signaling system, and protein export pathways, which support the PLD activation and signaling. The PLD signaling is also known to play a role in receptor endocytosis (Shen et al., 2001) and the endocytosis of synaptic vesicles in synaptosomes (Kobayashi and Kanfer, 1987). The receptor endocytosis of growth factor receptors such as EGFR is based on the PLD1 activation by PKCα and RALA proteins, both of which have been identified in this study.
However, the synaptic vesicle endocytosis is mediated by glutamatergic PLD activation, which explains the enrichment of endocytosis and glutamatergic synapse pathways in the hypothalamus. PLD signaling is also implicated in the synthesis of neurotransmitters such as acetylcholine and secondary messengers such as diacylglycerol (DAG). This ultimately assists in neurotransmitter activity, cholinergic signaling pathways, activation of PKC mediated calcium signaling for controlling glial cell proliferation, and long-term potentiation (Hattori and Kanfer, 1985; Lester and Bramham, 1993), all of which are represented among the enriched pathways identified from current data (Supplementary Table S12).
Interestingly, PLD also has a neuroprotective role in neurodegenerative diseases such as AD (Kanfer et al., 1986, 1996). We identified two isoforms of PLD in this study, PLD1 and PLD3 both of which are associated with the regulation of β-amyloid levels in the Alzheimer's brain (Cai et al., 2006; Demirev et al., 2019; Satoh et al., 2014). Several studies have elucidated the protective role of PLD1 in neurodegenerative disorders; however, the exact mechanism behind PLD3 activity toward preventing AD pathogenesis is still unclear. We observed that the PLD3 gene is expressed highest in the pituitary gland followed by the hypothalamus in the human brain as per the GTEx dataset. Thus, exploring the molecular mechanisms supporting the PLD3 activity specific to the hypothalamus and pituitary gland might lead to a better understanding of the pathophysiology of neurodegenerative disorders affecting these regions of the brain.
Conclusions
A deep proteomic analysis was performed in this study to catalog the normal human hypothalamic proteome, which has been missing for such an important organ so far. As a proof of concept, our study provided evidence that hypothalamic proteins can undergo several PTMs that may be required for normal hypothalamic functions. A proteogenomic analysis also resulted in the discovery of multiple novel protein-coding regions as we identified proteins from noncoding regions (pseudogenes) and proteins translated from sORFs that can be missed using the traditional pipeline of proteomics data analysis. In addition, we observed an enrichment of proteins pertaining to autophagy and adult neurogenesis in the proteome data.
Several proteins crucial to major regulatory functions of the hypothalamus were found to be associated with PLD signaling. Thus, the role of PLD signaling in the human hypothalamus demands further investigation. Furthermore, the exploration of the adult hypothalamic neurogenesis-related proteins identified in this study might open the possibility to better understand the metaplasticity of the human brain.
As a future perspective, extensive proteomic profiling of the hypothalamic nuclei would further elaborate on the role and functional characterization of several hypothalamus-specific proteins and pathways.
Footnotes
Acknowledgments
The authors thank the Department of Biotechnology, Government of India for research support to the Institute of Bioinformatics, Bangalore. O.C., L.G., and P.M. are recipients of Inspire Fellowship from the Department of Science and Technology (DST), Government of India. The authors also thank the Ministry of Health and Family Welfare, Government of India for providing instrumentation to NIMHANS-IOB laboratory, NIMHANS, Bangalore. NIMHANS-IOB facility is supported by DBT Program Support on Neuroproteomics and infrastructure for proteomic data analysis. The authors also thank Human Brain Bank Repository, NIMHANS, Bangalore, for providing human brain samples for the study.
The authors thank Yenepoya (Deemed to be University) for the access to mass spectrometry instrumentation at the Center for Systems Biology and Molecular Medicine (CSBMM). The authors thank Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka for the support to the CSBMM at Yenepoya (Deemed to be University) under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). The transcriptome data used for the correlation analyses with the proteomics data described in this article were obtained from the GTEx Portal on November 21, 2020.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for conducting this study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.