
Abstract
Multiomics study designs have significantly increased understanding of complex biological systems. The multiomics literature is rapidly expanding and so is their heterogeneity. However, the intricacy and fragmentation of omics data are impeding further research. To examine current trends in multiomics field, we reviewed 52 articles from PubMed and Web of Science, which used an integrated omics approach, published between March 2006 and January 2021. From studies, data regarding investigated loci, species, omics type, and phenotype were extracted, curated, and streamlined according to standardized terminology, and summarized in a previously developed graphical summary. Evaluated studies included 21 omics types or applications of omics technology such as genomics, transcriptomics, metabolomics, epigenomics, environmental omics, and pharmacogenomics, species of various phyla including human, mouse, Arabidopsis thaliana, Saccharomyces cerevisiae, and various phenotypes, including cancer and COVID-19. In the analyzed studies, diverse methods, protocols, results, and terminology were used and accordingly, assessment of the studies was challenging. Adoption of standardized multiomics data presentation in the future will further buttress standardization of terminology and reporting of results in systems science. This shall catalyze, we suggest, innovation in both science communication and laboratory medicine by making available scientific knowledge that is easier to grasp, share, and harness toward medical breakthroughs.
Introduction
Multiomics is an important research trend that integrates data obtained with two or more omics types such as genomics, transcriptomics, proteomics, metabolomics, epigenomics, miRNomics/ncRNomics, phenomics, and pharmacogenomics. Combining various omics layers enables researchers to analyze intricate biological big data.
There are different approaches to multiomics; some studies report their own experimental data (Huang et al., 2015), other studies are in silico where authors integrate previously published data (Cannistraci et al., 2013) and some combine in silico approach with their own experimental data (Mehla and Ramana, 2015). Owing to development of new technologies, such as next-generation sequencing (NGS), high-throughput DNA sequencing, RNA-seq, and advances in mass spectrometry, the use of multiomics is widespread in many scientific fields, including medicine, pharmacy, environment, and biological process development.
In medicine, for example, a multiomics study provided insights into miR-145-mediated tumor suppression that could also be used as a general strategy to study the targets of individual miRNAs (Huang et al., 2015). Furthermore, Mehla and Ramana (2015) used integrated in silico genome-wide approaches to discover novel drug targets for food-borne pathogen Campilobacter jejuni. Leading to nutrigenomics, Takahashi et al. (2014) integrated genomics, transcriptomics, proteomics, and metabolomics to discover metabolic alterations underlying the effect of coffee consumption. In addition, Pisithkul et al. (2019) performed a time-resolved analysis of the metabolome, transcriptome, and proteome, to better understand the process of biofilm formation in Bacillus subtilis.
Despite rapid advances, the multiomics field is facing challenges because of data incompleteness and fragmentation (Kunej, 2021). In our previous studies data integration from various sources in association with male reproduction, mastitis, mammary gland development, and milk traits (Cannistraci et al., 2013; Ogorevc et al., 2008; Ogorevc et al., 2009, 2011; Urh and Kunej, 2016; Urh et al., 2018), we were confronted with a large heterogeneity of data while extracting information from publications.
Our research interest has therefore advanced to proposing initiatives for standardized reporting in genomics. For instance, we have proposed a template for standardization of NGS data presentation (Pipan and Kunej, 2015), suggested a minimal checklist for reporting HIF-1α-target interactions (Slemc and Kunej, 2016) and reported an initiative for standardization of reporting genetics of male infertility and cryptorchidism (Traven et al., 2017; Urh and Kunej, 2016). Based on published genome-wide study approaches we organized omics approaches hierarchically and established a baseline for taxonomy of multiomics science and terminology development (Pirih and Kunej, 2017).
Researchers use at least 13 different synonyms for “multiomics,” which are presented in Table 1. Furthermore, data are scattered across various databases, research articles, and supplementary materials and are therefore in some cases not accessible for further research applications. However, this challenge can easily be diminished by standardization of reporting. A simple metadata omics checklist for a standardized approach in omics studies has already been proposed by Kolker et al. (2014). A supplement to the metadata checklist (Pirih and Kunej, 2018) with a graphical summary approach could enable researchers to define a multiomics study in a single figure.
Topline Analysis, Findings, and Outlook
An executive summary of this study.
Overview of Synonyms for Integrated Omics in Study Titles or Keywords
Thirteen different synonyms for integrated omics are presented along with the study title and reference.
Striving for optimal comprehension, standardization, and classification of omics and multiomics research, additional efforts are needed to systematize and control development of the multiomics field, especially since the increase of the publications related to this field is expected.
The aim of the present analysis was to investigate current trends in multiomics studies by evaluating examples of published articles that used an integrated omics approach. Therefore, we (1) carried out literature mining for examples of published multiomics articles from diverse research fields that investigated species of various phyla, extracted relevant information from the studies, and entered it in graphical summaries; (2) supplemented the missing data with standardized terminology and identification numbers regarding loci, species, study types, and phenotypes; and (3) created a network graph summarizing omics layers included in the evaluated multiomics studies.
Materials and Methods
We reviewed and evaluated 52 selected research articles that have been retrieved from the search engines PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and Web of Science (http://apps.webofknowledge.com/) published between July 2006 and January 2021, using keywords: omics, integrated omics, integrative omics, multiomics, integromics, and panomics. Articles were evaluated and compared according to four main criteria: (1) locus, including locus type and number of loci; (2) species, including the number and name of species, breeds, or populations, and number of samples or participants; (3) method, including omics type and experimental approach or study type; and (4) phenotype.
For each reviewed article, data were complemented with standardized terminology and identification numbers from relevant ontologies. For locus types and species, Sequence Ontology (SO) ID from the Sequence Ontology browser (www.sequenceontology.org) and Taxonomy ID from the Taxonomy Database (https://www-ncbi.nlm.nih.gov/taxonomy) were used, respectively. Phenotypes were supplemented with terminologies and ID numbers according to ontologies: Vertebrate Trait Ontology (VTO) (https://bioportal.bioontology.org/ontologies/VT), Plant Ontology (PO) (https://bioportal.bioontology.org/ontologies/PO), Disease Ontology (DO) (https://disease-ontology.org/), International Classification of Diseases (ICD) (https://icd.who.int/browse11/l-m/en), and Human Phenotype Ontology (HPO) (https://hpo.jax.org/app). Other phenotypes were obtained from specific ontologies at The Open Biological and Biomedical Ontology (OBO) Foundry (www.obofoundry.org).
Data with appropriate terminologies were summarized in a graphical summary introduced by Pirih and Kunej (2018). For visual presentation, a network graph of different omics layers in all reviewed studies was created using Cytoscape (www.cytoscape.org), an open source bioinformatics software platform (Shannon et al., 2003).
Results
To create an overview of the top trends in multiomics research, we evaluated 52 examples of published studies from the field. The workflow and executive summary of the study are presented in the Figure 1 and Box 1.
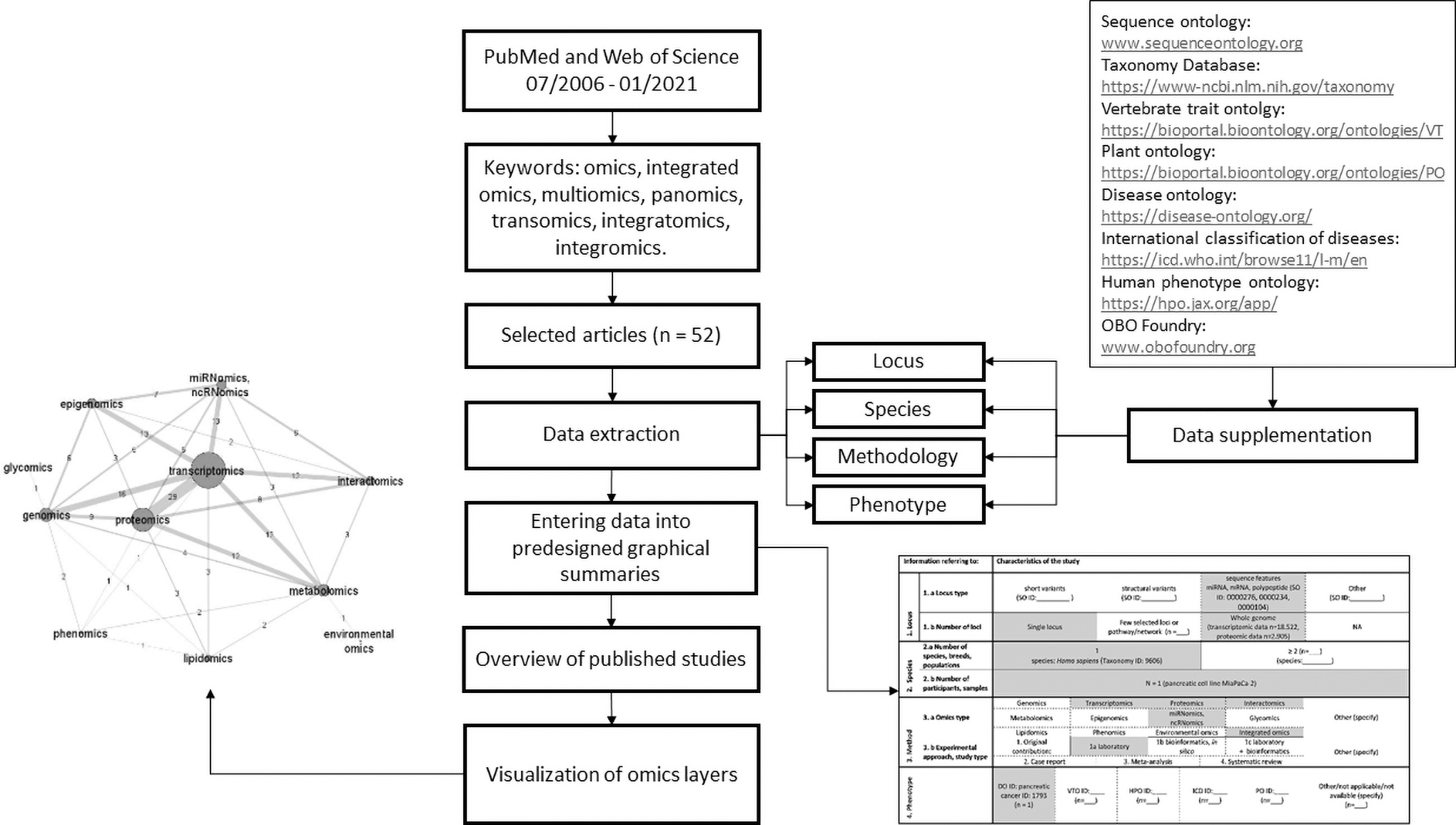
Graphical overview of the study. Workflow of the present study that includes a sample of a graphical summary and a network graph of omics layers in 52 evaluated studies.
The studies included altogether 21 different omics types or other applications of genomic technology, such as genomics, transcriptomics, epigenomics, and proteomics. The most frequently investigated phenotype and species were various types of cancer in human, but other traits, diseases, and biological processes in species of various phyla were also examined. Data synthesis from all evaluated studies revealed that the most recurrent integration of omics types was among transcriptomics and proteomics.
Literature mining, data extraction, and presentation in graphical summaries
Literature mining was performed to obtain a sample of published scientific articles that reported a multiomics approach. Fifty-two published articles combining two or more omics types were included in the present analysis (Table 2 and Supplementary Figs. S1–S50). Each study was evaluated regarding four main data types: (1) locus, (2) species, (3) methods, and (4) phenotype and summarized in a graphical abstract.
Overview of Omics Types in 52 Published Studies Using an Integrated Omics Approach

Gray squares signify the use of omics type (vertically) in a reviewed study (horizontally). Two, three, four, five and six omics types were integrated in 11, 22, 12, 5, and 2 studies, respectively. In the “Others” column, the applications of genomics technology or other information are presented with abbreviations. The number of analyzed species (vertically) varies from one to seven.
DGO, degradomics; MBO, microbiomics; MGO, metagenomics; MTO, metatransctriptomics; NGO, nutrigenomics; PGO, pharmacogenomics; PLO, phospholipidomics; PPO, peptidomics; RO, regulomics; SCO, secretomics; TGO, toxicogenomics.
(i) Types of investigated loci were curated according to SO and categorized into short variants (SNP and indel), structural variants (including copy number variation [CNV], deletion, sequence alteration), and other sequence features (including quantitative trait locus [QTL], epigenetically modified regions, histone modification), or other. Number of analyzed loci was retrieved from the evaluated studies and classified into three categories: a single locus study, a study investigating few selected loci, or the whole genome study. (ii) The second category of evaluated data included name of the species, and the number of analyzed species and samples.
(iii) Next, we obtained data on used methodology and categorized it by applied omics approaches in the studies and by experimental approach or study type, which could either be an original contribution, case report, meta-analysis, or systematic review. (iv) Investigated phenotypes were characterized as diseases or traits, and classified according to relevant ontologies.
The extracted information was entered into graphical summaries that had previously been developed by Pirih and Kunej (2018). Two examples of graphical summaries for evaluated studies using integrated omics approach are given in Figure 2. Figure 2A presents an evaluation of the study by Suzuki et al. (2014) in which a multiomics analysis of lung adenocarcinoma cells at genomic, transcriptomic, and epigenomic levels was conducted. Short variants (single nucleotide variant [SNV] and indel) as well as other sequence features (CNV, CpG island, methylated cytosine, histone modification, and mRNA) were investigated in human adenocarcinoma cell lines.
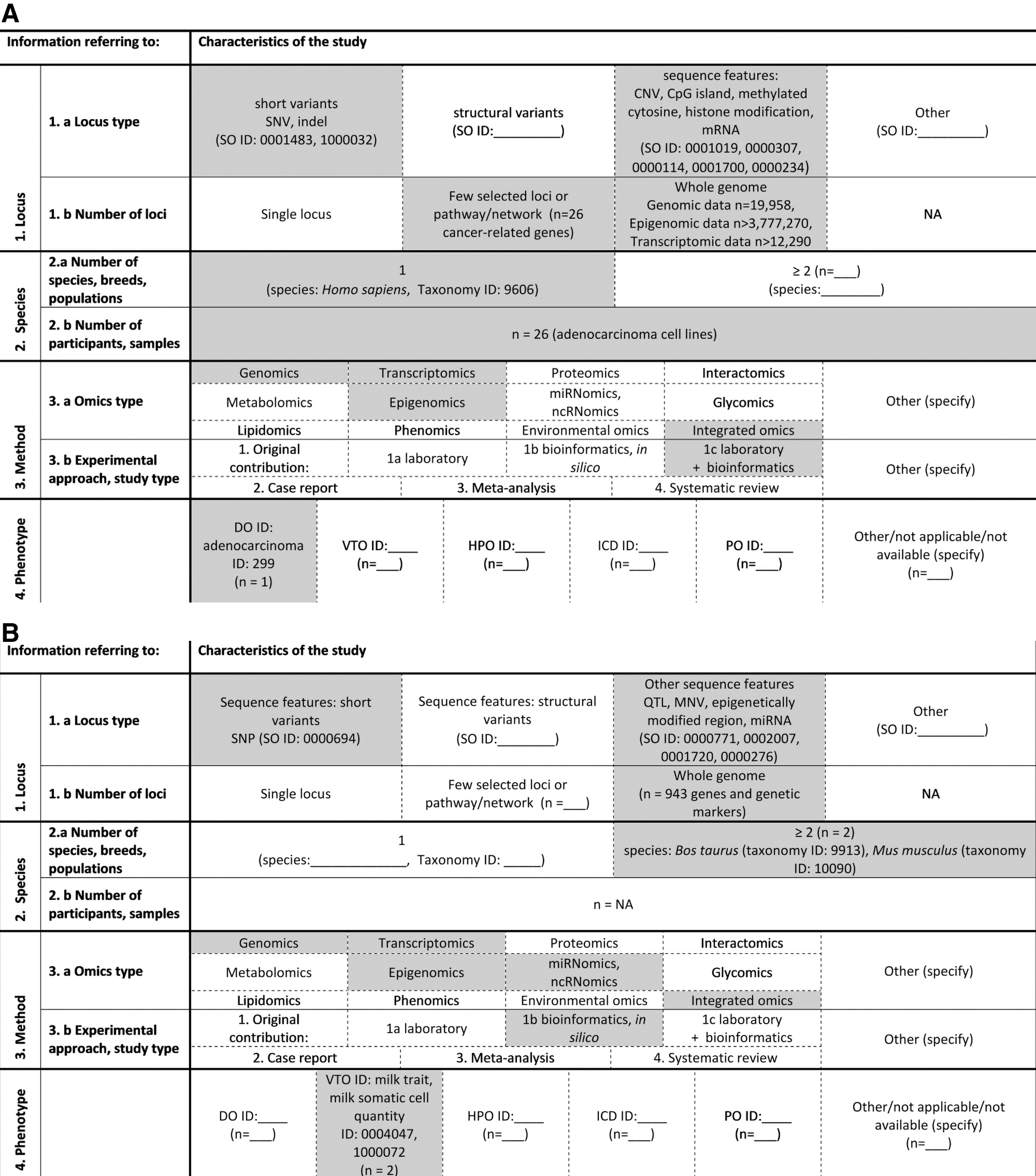
Graphical summary for an evaluated multiomics study.
Figure 2B presents a graphic summary of the in silico study by Ogorevc et al. (2009). In this integrative study of four omics layers, candidate loci in mammary gland development, milk production, and resistance or susceptibility to mastitis in cattle and mouse were investigated and additionally, a genetic map was developed, containing 943 sequence features: SNPs, QTLs, multiple nucleotide variants (MNVs), epigenetically modified regions, and miRNAs.
Complementation of extracted data from evaluated studies
Data complementation, curation, and streamlining of the retrieved data from evaluated studies was performed according to standardized terminology from relevant ontologies. For instance, in a study by Zhu et al. (2011) (Supplementary Fig. S18), aberration of DNA methylation, miRNA deregulation, and mRNA expression were analyzed. The terms “miRNA” (ID: 0000234) and “mRNA” (ID: 0000276) are available in the SO browser. The term “DNA methylation aberration” was streamlined to “silenced by DNA methylation” (ID: 0000895) and complemented with a parental term “epigenetically modified” (ID: 0000133).
Moreover, the number of investigated loci was obtained from the studies. If the number of analyzed loci in genome-wide studies was not explicitly reported, but the array or chip type applied in the research was provided, data regarding number of investigated loci were obtained online from specifications of the manufacturers, if available. For example, in a study by Takahashi et al. (2014) (Supplementary Fig. S3), Affymetrix GeneChip Mouse Genome 430 2.0 Array that profiles >39,000 gene transcripts was used for obtaining transcriptomic data. Some studies also performed a two-step analysis, for example, in a study by Calimlioglu et al. (2015) (Supplementary Fig. S5), a whole genome analysis was conducted to identify tissue-specific molecular biomarkers of type 2 diabetes and afterward, the top 20 reporter metabolites associated with type 2 diabetes were further explored as potential biomarkers.
Number of analyzed species, breeds, or populations were obtained from the studies. For instance, in a study by Kershaw et al. (2015), a single species was investigated. On the contrary, in a review by Aw and Fukuda (2015), the gut ecosystem was inspected and the number of species included in the study was uncountable. Furthermore, names of investigated species were complemented with Taxonomy ID numbers from The Taxonomy Database. For example, in the studies by Huang et al. (2015), Heydarian et al. (2014), Amiour et al. (2014), and Walther et al. (2010), human (Homo sapiens), mouse (Mus musculus), maize (Zea mais), and S. cerevisiae, with corresponding Taxonomy ID numbers 9606, 10090, 4577, and 4932, respectively, were investigated.
Information regarding phenotype was curated according to standardized terminology from relevant ontologies and supplemented with corresponding ID numbers. For example, Kikutake and Yahara (2016) investigated epigenetic biomarkers of lung adenocarcinoma. The phenotype is available both in DO and in ICD under the terms “lung adenocarcinoma” (ID: 2910) and “adenocarcinoma of bronchus or lung” (ID: 2C25.0), respectively. However, some phenotypes are not available in the proposed ontology databases. For instance, “cotton fibre development” (Wang et al., 2016) was curated according to PO as “plant tissue development stage” (ID: 0025423).
Overview of 52 evaluated studies that used a multiomics approach
Locus type and number of loci
Multiomics studies differed according to the locus type examined in the study. Evaluation of the studies revealed that types of loci were very heterogeneous, including either parent or child terms, and at least 32 different locus types were considered. The most investigated locus type was mRNA, included in 42 studies, followed by protein and miRNA, included in 29 and 14 studies, respectively.
Studies also reported other, very heterogeneous loci on various hierarchy levels. These include the following: SNP (ID: 0000694) (Bowler et al., 2013), SNV (ID: 0001483) (Suzuki et al., 2014), indel (ID: 1000032), and MNV (ID: 0002007) (Karničar et al., 2016), epigenetically modified region (ID: 0001720) (Girgis et al., 2012), histone modification (ID: 0001700) (Kikutake and Yahara, 2016), CpG islands (ID: 0000307) (Suzuki et al., 2014), QTL (ID: 0000771) (Ogorevc et al., 2011), CNV (ID: 0001019) and aneuploid chromosome (ID: 0000550) (Cannistraci et al., 2013), promoter (ID: 0000167) (Heydarian et al., 2014), ncRNA (ID: 0000655) (Joshi et al., 2020), protein–protein interaction (ID: 0001093) (Dimitrakopoulou et al., 2014), and others (Supplementary Figs. S1–S50). The hierarchies of the loci are presented in the SO browser.
Among the 52 evaluated studies, 48 (48/52) were performed on a whole genome scale. In three of the former (3/48), a whole genome analysis was combined with a single locus research (Bowler et al., 2013; Granadillo et al., 2020; Huang et al., 2015) and in eight cases (8/48) with a few selected loci or pathways (Ge et al., 2018; Mehla and Ramana, 2015; Nativio et al., 2020; Suzuki et al., 2014; Takahashi et al., 2014; Van Den Hof et al., 2014; Wang et al., 2009; Wang et al., 2016). The analysis of a few loci or pathways was performed independently in two studies (2/52) (Aw and Fukuda, 2015; Walther et al., 2010) and in two review studies, number of investigated loci was not applicable (2/52) (Joshi et al., 2020; Monte et al., 2014).
Species
The set of evaluated studies varied depending on organisms of interest and the number of participants or samples. Number of analyzed species, populations, or breeds extended from one to seven species, as given in Table 2.
Forty-two studies examined a single species and in 10 studies, two or more species were investigated, for example, in Cannistraci et al. (2013), Dimitrakopoulou et al. (2014), and Chen et al. (2016). Altogether, in 30 studies analyses were performed in human, 7 of those used a comparative cross-species approach, for example, Ogorevc et al. (2011), Aw and Fukuda (2015), and Vernocchi et al. (2020). In other studies various mammals were investigated, including mouse, cow, and pig, but the examined species also included plants, such as arabidopsis (Santoni et al., 2014), yeast (Kershaw et al., 2015), fungi (Culibrk et al., 2016), microbiota, and bacteria of various phylum (Vernocchi et al., 2020).
Methods according to omics type and experimental approach
Altogether, 52 studies used 21 classical omics approaches and applications of omics technology, including 10 classical omics types: genomics, transcriptomics, proteomics, interactomics, metabolomics, epigenomics, miRNomics/ncRNomics, glycomics, lipidomics, phenomics, environmental omics and integrated omics, as well as other omics types: metagenomics, metatranscriptomics, microbiomics, degradomics, peptidomics, regulomics, phospholipidomics, secretomics, and applications of omics technology: pharmacogenomics, toxicogenomics, and nutrigenomics. In Table 2, omics types included in the 52 studies are presented.
The most frequent omics type occurring in 47 studies is transcriptomics. Followed by proteomics, genomics (DNA level), metabolomics, epigenomics, and miRNomics/ncRNomics, used in 31, 18, 16, 13 and 13 studies, respectively. Phenomics (Joshi et al., 2020; Monte et al., 2014) and lipidomics (Badoud et al., 2017; Clish et al., 2004; Joshi et al., 2020; Overmyer et al., 2021) were reported in two and four studies, respectively, whereas environmental omics (Aw and Fukuda, 2015) and glycomics (Ge et al., 2018) were each applied in one study. The number of integrated omics types in the studies varied from two to six. The most recurrent synthesis of two omics types was among transcriptomics and proteomics, included in 29 studies, followed by integration between transcriptomics and genomics, included in 16 studies.
Considering methodology, studies also differed according to study type. Forty-four reviewed studies were original contributions; however, there were also 4 systematic reviews (Aw and Fukuda, 2015; Culibrk et al., 2016; Joshi et al., 2020; Monte et al., 2014), 2 meta-analysis (Gov et al., 2017; Roume et al., 2015), and a case report (Granadillo et al., 2020). Among the 44 original contributions, studies differed regarding experimental approach. Nine evaluated studies (9/44) used a molecular genetics (“wet” lab) approach. Nonetheless, 14 studies (14/44) were performed in silico, where authors used or retrieved information from databases or previously published data, or used bioinformatics (“dry” lab) approach. Finally, 21 (21/44) original contributions combined laboratory approach with bioinformatics.
Phenotype
According to studied phenotype, studies differed in whether they investigated a disease, a physiological mechanism or a biological process, a trait, or other feature. The most commonly presented phenotypes in the evaluated studies were diseases of which the most abundant were various types of cancer, occurring in 13 studies, for example, in Pan et al. (2009), Zhu et al. (2011), and Nymark et al. (2011), followed by diabetes, investigated in 4 studies (Aw and Fukuda, 2015; Calimlioglu et al., 2015; Ge et al., 2018; Monte et al., 2014), and cardiovascular diseases in 3 studies (Badoud et al., 2017; Clish et al., 2004; Joshi et al., 2020). Two of the selected studies also investigated the coronavirus disease 2019 (COVID-19) (Overmyer et al., 2021; Su et al., 2020).
As many as 13 studies examined biological processes or physiological mechanisms, such as cell development (Heydarian et al., 2014), tolerance to chemicals (Yang et al., 2012a), and biofilm formation (Suzuki et al., 2014). Other phenotypes included traits, for example, milk trait (Ogorevc et al., 2008) and behavioral circadian rhythm (Wang et al., 2009).
An overview of omics types and number of investigated species in all evaluated studies is given in Table 2. Furthermore, the data on applied omics approaches from Table 2 are presented as a network graph in Figure 3. The visualization depicts omics approaches most often applied together in the evaluated multiomics studies. Each line represents an integration between two omics types. The numbers on the edges and the thickness of the edges represent the frequency of merging two omics types, whereas the size of a circle denotes the number of studies using a certain omics approach.
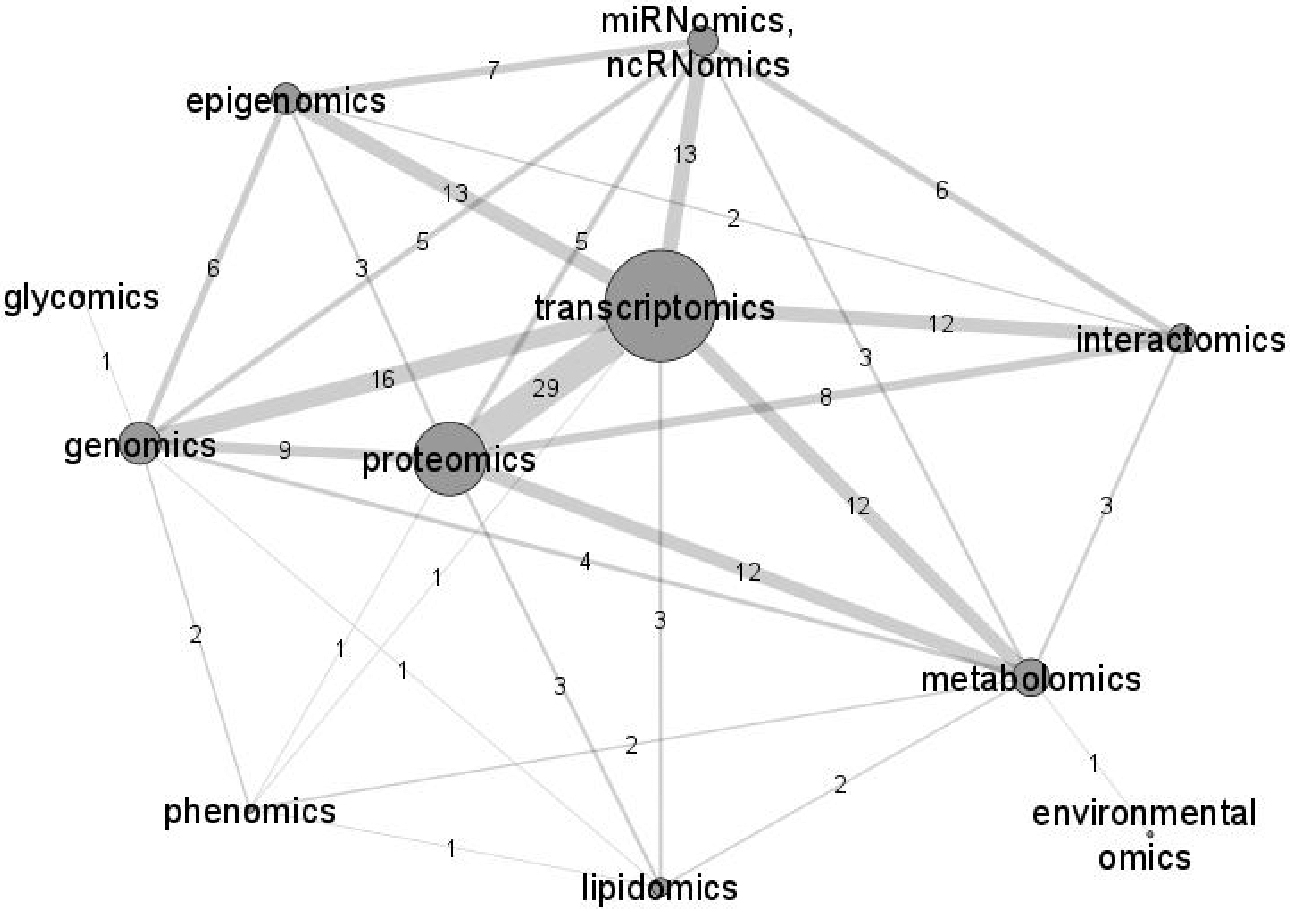
A presentation of connections between omics types in 52 evaluated studies. Each line represents a synthesis of two omics types in an integrated omics study. The width of an edge and a number on the edge represent the number of connections between two omics types in different studies. The size of the node denotes the number of studies that used the omics approach.
Discussion
In this study, we performed an analysis of the top trends in multiomics research. We applied a previously proposed classification scheme and a graphical summary for comprehensive presentation of omics studies (Pirih and Kunej, 2017, 2018). Analysis of 52 multiomics studies revealed a heterogeneity of the field, absence of terminology, and standardized results presentation.
In the evaluated set of studies, >32 different locus types were investigated and analyses were performed in various species: human and other mammals, plants, fungi, yeast, and bacteria. According to methodology, 14 studies were performed utterly in silico, 9 studies used a wet lab approach, and 21 studies combined both approaches. Further growth of bioinformatics studies is yet to be expected owing to the increase in the complex data attained in multiomics research. Similarly, full-genome sequencing methods are continuously developing, with >380 NGS methods already available (http://enseqlopedia.com).
Although the field of multiomics is rapidly advancing, it is impossible to determine the exact number of all integrative omics studies as some publications do not term “multiomics” in the title or keywords. Some earlier studies were published before this term became universally used in research and thus do not include “multiomics,” although they contain two or more omics types (Ogorevc et al., 2008). However, in some studies, synonyms for multiomics, such as “integratomics” (Cannistraci et al., 2013; Maver and Peterlin, 2011), or multiple individual omics types, such as transcriptomics, proteomics, and metabolomics (Pisithkul et al., 2019) are used.
Data extraction regarding the locus types and number of loci investigated from published studies is demanding. Not all locus types are yet available in databases and possess identification numbers, for example, “DNA methylation aberration” is often used in the studies but is not included in the SO yet (Zhu et al., 2011). Thus, in the present analysis, it was not possible to discern the exact number of different locus types investigated in the evaluated studies.
Moreover, the terminology regarding some locus types is not yet entirely developed and organized. For instance, delineation between SNPs and SNVs is not always evident. In addition, many multiomics studies do not investigate DNA loci, mRNA, or proteins, but rather focus on other levels of expression and metabolism, such as lipids or metabolites. For better classification, ontologies have to be extended to also involve other molecules in the future. Furthermore, the minimal number of investigated loci, which is still defined as a “whole genome” study, is yet to be specified. For example, in an in silico study by Ogorevc et al. (2009), 943 candidate loci were assembled, however, these loci are widespread across the entire genome and therefore, we classified the study as a “whole genome” study.
Data curation concerning species can be challenging. In some studies, number of species or samples is not specified and is sometimes even irrelevant. For instance, in studies researching microbial communities or gut ecosystems, the number of included populations is unobtainable, because of the complexity of the analysis. However, in a review study by Aw and Fukuda (2015), the gut ecosystem was explored and the authors provided the four major phyla, included in the research (Supplementary Fig. S2).
Concerning categorization of omics types, some authors had already classified used methods into separate omics layers. For example, mRNA expression profiling using a microarray hybridization can be classified as transcriptomics (Van Den Hof et al., 2014). Similarly, we assigned the omics types according to the methods used in the evaluated studies. This is challenging because of the fact that definitions for individual omics types are not yet set, as there is no classification system of methods according to omics layers. Moreover, the number of studies using variations and combinations of classical omics types, such as metagenomics/metatranscriptomics (Aw and Fukuda, 2015), toxicogenomics (Lv et al., 2012), and proteogenomics (Konc et al., 2017), is growing.
Similar difficulty exists with information regarding phenotype. Although some phenotypes such as various types of cancer or diseases are accessible in the ontologies, terms describing a specific response in the body are more challenging to classify. The discrepancies in trait nomenclature have already been discussed by Park et al. (2013) along with the examples of proposed adjustments to phenotype terms or traits using VTO. Moreover, different ontology databases partially overlap, for example, “obesity,” “chronic kidney disease,” “multiple sclerosis,” and others are accessible both in DO and in ICD databases.
Our study also has limitations, such as the limited size of the sampled published multiomics articles. To gain even more in-depth insight into the rapidly evolving field of multiomics, an evaluation of additional studies is necessary. Moreover, interpretation of results in the evaluated studies is challenging owing to the incompleteness of data and lack of standard terms. For instance, in some studies, the type of diabetes is irrelevant and is therefore not specified; however, this information is important for further genotype–phenotype association studies.
The field of multiomics itself is very diverse, as it already enables researchers to unravel the conundrums of various processes including microbial, plant, and mammalian physiology and cell differentiation (Wang et al., 2016), increase industrial yield by yeast improvement in biotechnology (Karničar et al., 2016), develop biomarkers for human cancer (Girgis et al., 2012), and underlie the mechanisms of circadian rhythm (Wang et al., 2009). Multiomics approach is also applicable for understanding pathophysiology in COVID-19, identifying potential therapeutic opportunities, and determining severity of the disease in early stages of infection (Overmyer et al., 2021; Su et al., 2020). Moreover, in the next decade, the multiomics field is expected to evolve even further as discussed in Kunej (2021).
Nonetheless, some challenges still need to conquered, such as introduction of reporting standardization, which will encourage researchers to consider the FAIR Guiding Principles (findability, accessibility, interoperability, and reusability) (Wilkinson et al., 2016). In most articles a part of the information was obfuscated by lack of standard terms, which indicates that standardization of reporting data is an urgent task. Hence, a graphical summary is a promising aid to future researchers and readers, as it enables consistent and dependable presentation of results with standardized terminology.
Conclusions and Future Directions
This study is an overview of current top trends in multiomics research based on evaluation of 52 published studies. Each of the evaluated articles was presented in a form of a graphical abstract, including data referring to locus, species, methodology, and phenotype. In addition, this study will contribute to raise the researchers' awareness of standardized reporting in the multiomics field. Evaluation of additional studies would further enhance our insight into the state of multiomics, enable standardization of reporting, and lead to more coordinated development of the field.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.