
Abstract
Maturity-onset diabetes of the young (MODY) is a highly heterogeneous group of monogenic and nonautoimmune diseases. Misdiagnosis of MODY is a widespread problem and about 5% of patients with type 2 diabetes mellitus and nearly 10% with type 1 diabetes mellitus may actually have MODY. Using next-generation DNA sequencing (NGS) to facilitate accurate diagnosis of MODY, this study investigated mutations in 13 MODY genes (HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, and KCNJ11). In addition, we comprehensively investigated the clinical phenotypic effects of the genetic variations identified. Fifty-one adult patients with suspected MODY and 64 healthy controls participated in the study. We identified 7 novel and 10 known missense mutations localized in PDX1, HNF1B, KLF11, CEL, BLK, and ABCC8 genes in 29.4% of the patient sample. Importantly, we report several mutations that were classified as “deleterious” as well as those predicted as “benign.” Notably, the ABCC8 p.R1103Q, ABCC8 p.V421I, CEL I336T, CEL p.N493H, BLK p.L503P, HNF1B p.S362P, and PDX1 p.E69A mutations were identified for the first time as causative variants for MODY. More aggressive clinical features were observed in three patients with double- and triple-heterozygosity of PDX1-KLF11 (p.E69A/p.S182R), CEL-ABCC8-KCNJ11 (p.I336, p.G157R/p.R1103Q/p.A157A), and HNF1B-KLF11 (p.S362P/p.P261L). Interestingly, the clinical effects of the BLK mutations appear to be exacerbated in the presence of obesity. In conclusion, NGS analyses of the adult patients with suspected MODY appear to be informative in a clinical context. These findings warrant further clinical diagnostic research and development in different world populations suffering from diabetes with genetic underpinnings.
Introduction
Maturity-onset diabetes of the young (MODY) accounts for 1–2% of all diabetes cases in the majority of the populations studied (Nkonge et al., 2020; Urakami, 2019). However, misdiagnosis of MODY is a widespread problem and about 5% of patients with type 2 diabetes mellitus (T2DM) and nearly 10% with type 1 diabetes mellitus (T1DM) may actually have MODY (Covantev et al., 2016).
MODY is a monogenic disorder characterized by autosomal dominant inheritance in at least two generations, beta cell dysfunction, and early-onset diabetes in adolescence or young adulthood without evidence of insulin resistance such as obesity and/or acanthosis nigricans (Juszczak and Owen, 2014; Nkonge et al., 2020; Urakami, 2019). MODY represents genetic, metabolic, and clinical heterogeneity, depending on genetic etiology (Gardner and Tai, 2012). Although with some exceptions, MODY patients typically have nonprogressive, mild, stable fasting hyperglycemia and respond to oral diabetes medications such as sulfonylurea (SU) (Urakami, 2019). Patients have measurable C-peptide levels in the presence of hyperglycemia and have normal triglycerides and/or normal or elevated high-density lipoprotein cholesterol (HDL-C) levels (Covantev et al., 2016).
MODY develops with the heterozygous alterations leading to pancreatic beta cell defects such as impaired glucose-stimulated insulin secretion in at least 14 genes (Table 1), most of which are transcription factors, hepatocyte nuclear factor 4α (HNF4A; MODY1), glucokinase (GCK; MODY2), hepatocyte nuclear factor 1α (HNF1A; MODY3), pancreatic and duodenal homeobox 1/insulin promoter factor 1 (PDX1/IPF1; MODY4), hepatocyte nuclear factor 1β (HNF1B; MODY5), neurogenic differentiation 1 (NEUROD1; MODY6), Kruppel-like factor 11 (KLF11; MODY7), carboxyl ester lipase (CEL; MODY8), paired-box-containing gene 4 (PAX4; MODY9), insulin (INS; MODY10), B lymphocyte kinase (BLK; MODY11), ATP-binding cassette transporter subfamily C member 8 (ABCC8; MODY12), potassium channel, inwardly rectifying subfamily J, member 11 (KCNJ11; MODY13), and adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper containing 1 (APPL1; MODY14) (Nkonge et al., 2020; Prudente et al., 2015).
The Causative Genes for Maturity-Onset Diabetes of the Young Subtypes
ATP, adenosine triphosphatase; HbA1c, glycosylated hemoglobin A1c; K-ATP, potassium-adenosine triphosphatase; MODY, maturity-onset diabetes of the young; SUR1, sulfonylurea receptor 1; T1DM, type 1 diabetes mellitus.
Genes associated with subtypes MODY1–6 were defined as “causative,” whereas eight genes associated with subtypes MODY7–14 were defined as “probably causative” (Nkonge et al., 2020).
Today, the diagnosis of MODY should be more carefully differentiated because of the increasing prevalence of diabetes, the frequent family history of diabetes among individuals with diabetes, and the younger age at onset of diabetes. In addition to the 14 known responsible genes, as well as the unidentified subtype(s) of the responsible gene defined as MODY X, misdiagnosis due to clinical features overlapping with T1DM and T2DM further increases the complexity of the MODY clinic.
The definitive diagnosis of MODY can only be made by detecting mutations in the genes responsible for MODY using DNA sequencing. More comprehensive genome screening studies are needed in our country to more precisely determine the prevalence and subtypes of MODY. An accurate diagnosis will help to apply appropriate treatment and to identify the risk among family members.
The aim of this study was to identify MODY-specific gene variations in 13 genes (except APPL1) associated with MODY in adult patients with a clinical suspicion of MODY by targeted next-generation DNA sequencing (NGS), and to diagnose MODY subtypes with an integrated analysis by examining genetic, bioinformatic, clinical, and biochemical data together.
Materials and Methods
Characteristics of the study groups
Fifty-one adult patients (aged 23–64 years) with clinical suspicion of MODY and 64 adult healthy controls (aged 24–66 years) were enrolled in this study (so-called MODY-IST-Adult) from Istanbul University, Istanbul Faculty of Medicine, Department of Internal Medicine in Turkey. All patients and controls were subjected to a detailed physical examination and biochemical analysis. The patient group was chosen according to clinical parameters, including age at onset of diabetes between 16 and 40 years, family history of diabetes in a minimum of two generations suggesting an autosomal dominant inheritance in pedigree analysis, preserved endogenous insulin reserve (fasting serum C-peptide ≥0.6 ng/mL); absence of ketoacidosis, no severe microvascular complications plus negative pancreatic autoantibodies (islet cell cytoplasmic, or islet antigen 2, or glutamic acid decarboxylase antibody).
Healthy subjects without any metabolic disease and no family history of diabetes in first-degree relatives were included in the control group during a routine examination.
Written informed consent was obtained from all patients and the study was approved by the Institutional Review Board (Istanbul Faculty of Medicine Ethical Committee: 06.20.2014/922). Clinical parameters (age, sex, body mass index [BMI], medications, chronic diseases, medical interventions, etc.) were recorded using a patient follow-up form. This study was conducted in accordance with the Declaration of Helsinki.
DNA purification and library preparation
Genomic DNA was isolated from peripheral blood samples taken during routine visits using the Epicenter MasterPure™ (Lucigen, WI, USA) DNA purification kit. DNA purity and concentration measurements were performed spectrophotometrically using NanoDrop 2000c (ThermoFisher Sci., MA, USA). Extracted DNA was quantified using the Qubit™ dsDNA HS Assay Kit and the Qubit® 3.0 Fluorometer (Invitrogen, CA, USA).
Exons and exon/intron boundaries of 13 MODY-related genes, including HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, KCNJ11, and ABCC8, were amplified through long polymerase chain reaction using Thermal Cycler (C1000; Biorad, USA). Since the APPL1 gene has not yet been associated with MODY at the time of application for project funding, it was not included into the panel. Libraries were constructed using the TruSeq® Custom Amplicon v1.5 Exome Library Prep kit (Illumina, Inc., San Diego, CA, USA).
Exome sequencing and NGS data analysis
Paired-end sequencing of the libraries was performed on MiSeq 4000 System (Illumina, Inc.). An average sequencing depth of 221 thousand reads per sample was obtained resulting in an average of 90% of targeted bases that covered at least 10 × .
Analysis of NGS data was performed according to the pipeline developed in our previous study (Demirci et al., 2021). Briefly, all samples were analyzed according to the Genome Analysis ToolKit (GATK) best practices pipeline (Van der Auwera et al., 2013). The human genome build37 (GRCh37/hg19) was used as the reference genome (http://genomereference.org). The status, novelty, functional impact, and pathogenicity of variants were assessed, and annotated variants with an overall population frequency of <5% were filtered out and compared between study groups. The Hardy/Weinberg equilibrium (HWE) was tested for each variant, and relative risk (RR) was determined. Visualizations were performed using the R packages cBioPortalData v2.2.8 (Ramos et al., 2020) and forest plot v1.10.1 (https://cran.r-project.org/web/packages/forestplot). The MutationMapper tool of cBioPortal was used to map mutations on proteins and their domains (Gao et al., 2013).
Statistical analyses
Statistical analyses were performed with SPSS v.20.0 (IBM SPSS, Inc., Chicago, IL, USA). Quantitative variables were presented as means and standard deviations, and Student's t-test was used for comparison between two groups. Categorical variables were presented as numbers and percentages, and statistical analyses between the study groups were performed using the chi-square test. p Values <0.05 were considered statistically significant.
Results
Clinical investigation
A comparative analysis of demographic, clinical, and biochemical characteristics of the study groups (Table 2) revealed significant differences in BMI (p = 0.001), fasting blood glucose (FBG) (p = 0.001), glycosylated hemoglobin A1c (HbA1c) (p = 0.001), triglycerides (p = 0.003), HDL-C (p = 0.013), alanine aminotransferase (p = 0.021), high-sensitive C-reactive protein (p = 0.001), thyroid stimulating hormone (p = 0.027), free thyroxine (p = 0.004), white blood cells (p = 0.001), and family history of diabetes (p = 0.001). All parameters, except HDL-C, were higher in the MODY group than control group.
Characteristics of the Study Groups
Bold p values indicate statistical significance.
BMI, body mass index; FBG, fasting blood glucose; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein-cholesterol; VLDL-C, very low-density lipoprotein cholesterol; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma glutamyl transferase, hs-CRP, high-sensitive C-reactive protein; TSH, thyroid stimulating hormone; FT4, free thyroxine; ACR, urine albumin-to-creatinine ratio; BUN, blood urea nitrogen; e-GFR, estimated glomerular filtration rate.
The mutation loads of MODY genes
Paired-end sequencing (with an average length of 150 bp) of the exons and exon/intron boundaries (±100 bp) of 13 MODY-related genes resulted in an average sequencing depth of 221 thousand high-quality reads per sample, leading to an average of 90% of targeted bases that covered at least 10 × . Following the GATK best practice pipeline and using the human genome build37 (GRCh37/hg19) as the reference genome, we identified 135 genetic variations in 13 MODY-related genes (Figs. 1 and 2).

Mutations identified in MODY genes: ABCC8, BLK, CEL, GCK1, and HNF1A. MODY, maturity-onset diabetes of the young.
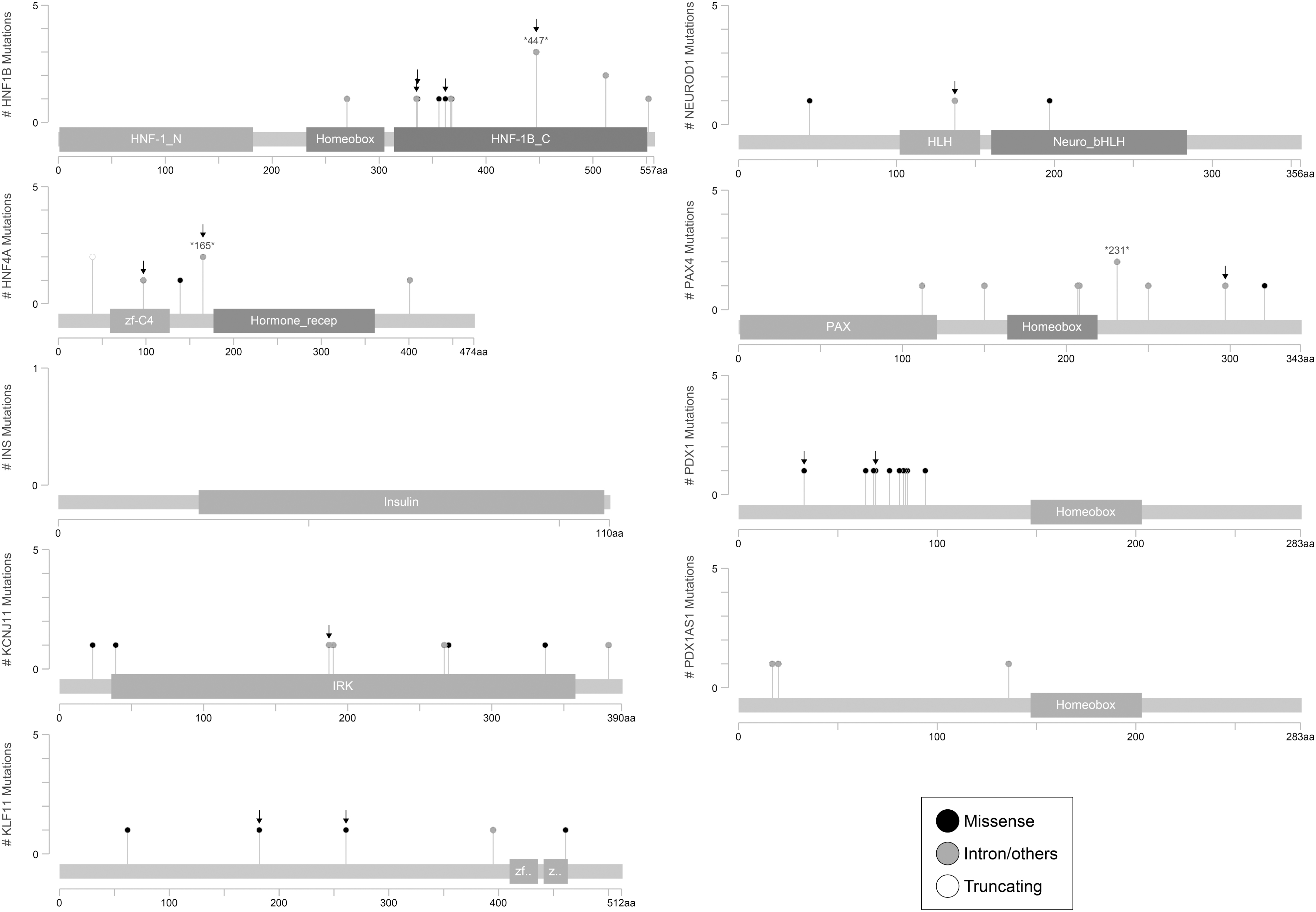
Mutations identified in MODY genes: HNF1B, HNF4A, INS, KCNJ11, KLF11, NEUROD1, PAX4, PDX1, and PDX1AS1.
Different mutation loads were observed in the MODY genes. The highest number of variations was observed in the ABCC8 gene (n = 30), followed by the HNF1A (n = 17), BLK (n = 15), and CEL (n = 15) genes. Ten variations each were detected in the HNF1B and PDX1 genes, and eight variations each in the KCNJ11 and PAX4 genes. The fewest variations were detected in the genes GCK (n = 6), HNF4A (n = 5), KLF11 (n = 5), NEUROD1 (n = 3), and PDX1-AS1 (n = 3). No variations in the gene INS were detected in the studied population.
Annotated variants in MODY genes
Among the identified variations, 41 variations were MODY-specific, that is, they were detected only in MODY patients compared with the control group. No MODY-specific mutation was detected in the HNF1A and INS genes. MODY-specific variants were analyzed in terms of their genomic features, pathogenicity, functional consequences, and literature ClinVar status (Tables 3 and 4).
Other Mutations Specific to Maturity-Onset Diabetes of the Young-Adult Group
Missense Mutations Specific to Maturity-Onset Diabetes of the Young-Adult Group
CI, confidence interval; HWE, Hardy/Weinberg equilibrium; NR, not reported.
MODY-specific missense mutations were identified in six genes, namely ABCC8 (n = 2), BLK (n = 4), CEL (n = 4), HNF1B (n = 2), KLF11 (n = 3), and PDX1 (n = 2) (Table 3). Among these, seven novel mutations, namely BLK p.L503P, ABCC8 p.R1103Q, ABCC8 p.V421I, CEL I336T, CEL p.N493H, HNF1B p.S362P, and PDX1 p.E69A, were identified for the first time in this study, whereas the remaining nine missense mutations were previously associated with MODY subtypes in ClinVar. The p.G157R and I488T mutations in CEL, the p.H336D mutation in HNF1B, the p.R461Q mutation in KLF11, and the p.P33T mutation in PDX1 have been described in the literature as MODY-associated pathogenic variants.
Moreover, we identified 4 synonymous variants in ABCC8, HNF1B, NEUROD1, and KCJN11; 19 intronic variants in ABCC8 (n = 9), BLK (n = 4), GCK (n = 2), HNF1B (n = 1), HNF4A (n = 2), and PAX4 (n = 1) in addition to a 3′UTR variant in HNF4A (Table 4). The synonymous mutations (i.e., ABCC8 c.334 C>T, NeuroD1 c.411 G>A, and KCNJ11 c. 561 G>A) were also identified for the first time in this study.
Double heterozygosity of PDX1-KLF11 (p.E69A/p.S182R), CEL-ABCC8-KCNJ11 (p.I336, p.G157R/p.R1103Q/A157A), and HNF1B-KLF11 (p.S362P/p.P261L) was observed in three patients (Table 5).
Clinical and Genetic Characteristics of Maturity-Onset Diabetes of the Young Subtypes in the Analyzed 13 Maturity-Onset Diabetes of the Young Genes
DPP-4i, dipeptidyl peptidase-4 inhibitor; GDM, gestational diabetes mellitus; N/A, not applicable; SU, sulfonylurea.
Considering that heterozygous MODY mutations in the transactivation, DNA-binding or dimerization domains of proteins, particularly transcription factors, lead to decreased gene expression and activity via haploinsufficiency (Ellard and Colclough, 2006), and nonsense, frameshift, and splicing mutations lead to mRNAs with premature stop codons in the channel- and enzyme-encoding genes (Colclough et al., 2013), we investigated which protein domains the mutations correspond to (Figs. 1 and 2) and whether amino acid substitution affects protein structure and function by estimating the likelihood of pathogenicity of missense mutations. The majority of the missense mutations were predicted to be deleterious or damaging, whereas four mutations, namely KLF11 p.P261L, BLK p.R450H, BLK p.L503P, and ABCC8 p.R1103Q, were classified as tolerated or benign (Table 3).
The RRs changed in the range of 2.27 (1.85–2.78) to 2.29 (1.87–2.82) for all variants, suggesting that MODY risk increased with the presence of variation. In addition, violations of HWE assumptions were observed for most MODY-specific variants (p > 0.05) (Tables 3 and 4).
Discussion
MODY comprises a heterogeneous group of monogenic disorders characterized by early-onset diabetes, pancreatic β cell dysfunction, and autosomal dominant inheritance. Our study of this large series of MODY gene mutations in 51 unrelated patients with a clinical suspicion of MODY once again underscores the substantial genetic heterogeneity of MODY. We identified 41 MODY-specific genetic variations, including 17 missense mutations, 4 synonymous mutations, 19 intronic variants, and one 3′UTR variant, in 13 MODY causative genes.
Seven novel and 10 known missense mutations localized in PDX1, HNF1B, KLF11, CEL, BLK, and ABCC8 genes were detected in 29.4% (n = 15) of MODY patients. Among them, p.E69A and p.P33T mutations in PDX1, p.S362P and p.H336D mutations in HNF1B, p.S182R and p.R461Q mutations in KLF11, p.I336T, p.N493H, p.G157R, and p.I488T mutations in CEL, p.H293Q and p.A71T mutations in BLK, and p.V421I mutation in ABCC8 were predicted to be deleterious or damaging. The missense mutations in KLF11 p.P261L, BLK p.R450H, BLK p.L503P, and ABCC8 p.R1103Q were predicted to be benign.
To the best of our knowledge, ABCC8 p.R1103Q, ABCC8 p.V421I, CEL I336T, CEL p.N493H, HNF1B p.S362P, and PDX1 p.E69A mutations were identified in MODY patients for the first time in this study. Nine of the patients were diagnosed as “MODY X” due to the absence of causative gene mutations in the analyzed MODY genes. Of the remaining patients, 4 were evaluated as T1DM, 22 as T2DM, and 1 as latent autoimmune diabetes in adults. More aggressive clinical features were observed in three patients with double heterozygosity of PDX1-KLF11 (p.E69A/p.S182R), CEL-ABCC8-KCNJ11 (p.I336T, p.G157R/p.R1103Q/p.A157A), and HNF1B-KLF11 (p.S362P/p.P261L) (Supplementary Table S1).
In the present study, three HNF4A variations (two intronic and one 3′UTR) were detected. One of the intronic variants (c.225–25T>A, rs200071662) was associated with T1DM in a North American cohort (Bennett et al., 2015); however, the other novel intronic variant (c.493–54G>A) and 3′UTR variant (c.*42C>T, rs965250768) were identified in this study and associated with MODY. As in the pediatric/adolescent MODY patients of our previous study (Demirci et al., 2021), we did not find any MODY-specific missense mutation in the HNF4A gene in the adult MODY patients of this study. On the contrary, we detected the HNF4A c.350 C > T p.T117I (rs1800961) missense mutation, which was previously detected in MODY studies in Turkey (Agladioglu et al., 2016; Karaoglan and Nacarkahya, 2021). However, since we also observed the p.T117I mutation in healthy controls, we did not evaluate it as a MODY-specific mutation in this study.
Heterozygous inactivating
In this study, two novel intronic GCK variations, c.-35C>A and c.867–75C>T, were identified, which are localized in intron 1 and intron 7, respectively. However, none of the MODY patients had the missense GCK mutation.
Heterozygous
In fact, many known (p.I27L and p.A98V) and novel (p.I404T, p.T544P, p.T547P, p.F543S, p.S550P, p.L546P, p.S553P, and p.T564P) missense mutations in the HNF1A gene were detected in our study group. However, they were not specific to identify MODY, since they were also observed in the healthy controls. Among these mutations, novel missense p.S553P (n = 2) and p.T564P (n = 1) mutations were detected only in healthy controls.
Among these missense mutations, we identified only p.Pro33T and p.E69A, which were predicted to be deleterious by bioinformatic tools, as being specific for MODY. Both mutations are localized in the transactivation domain of the PDX1. These mutations cause downregulation of the PDX1-bound genes, including the transcription factors MNX1 and PDX1, and insulin resistance gene CES1, reducing the differentiation efficiency of pancreatic progenitors and ultimately impairing beta cell differentiation and function (Wang et al., 2019). This study was the first to identify the PDX1 p.E69A missense mutation in MODY patients. Based on the in silico prediction algorithms, SIFT, and PolyPhen-2, the variant, p.E69A (c.A206C), is considered to be pathogenic.
The female patient (DAH33) who carried the PDX1 p.E69A mutation also carried the missense mutation KLF11 p.S182R and under insulin therapy. She was overweight and suffered from polycystic ovary syndrome, hypercholesterolemia, and menstrual irregularity. Her clinical features support a double heterozygosity for MODY4/MODY7. The patient with the PDX1 P33T mutation was a 26-year-old woman (DAH63) receiving insulin therapy. The patient had high FBG, HbA1c, and serum lipid levels, and low liver aminotransferase levels. In this patient, only the PDX1 mutation was present in the genes we examined, and she had obesity and a history of GDM. The PDX1 P33T mutation was first described in a large Italian family study by Gragnoli et al. This mutation has been reported to be associated with MODY4 and GDM phenotypes (Gragnoli et al., 2005). The clinical features of our patient with the p.P33T mutation are consistent with the findings of Gragnoli.
After the first report of
In our study group, five of the detected missense mutations of HNF1B gene were novel (p.G356E, p.H368P, p.T376P, p.S7P, and p.S362P) and one of them (p.H336D) was already presented in the dsSNP database. Among these, only p.H336D and p.S362P were MODY-specific mutations. These two mutations have been previously reported in studies conducted in the United States, Serbia, and Germany (Karges et al., 2007; Komazec et al., 2019; Thomas et al., 2008). The p.H336D mutation, in which the histidine located in the activation domain of the Hnf1β protein is replaced by an aspartic acid (Weber et al., 2006), was found in only one patient (DAH24).
A 35-year-old female patient with clinical features of young-onset diabetes (10 years of age), previous severe obesity, GDM, early cataracts, neuropathy, and nephropathy was compatible with the MODY5 phenotype. The male patient with p.S362P mutation (DAH62) also carried the heterozygous KLF11 p.P261L missense mutation. The patient received antidiabetic therapy, and had high FBG, and normal insulin/C-peptide levels, hypercholesterolemia, and hypertension. He had elevated aminotransferases without any other signs of liver dysfunction and a slightly increased urine albumin-to-creatinine ratio (49.5 mg/g). His clinical features support a double heterozygosity for MODY5/MODY7.
In this study, we detected the HNF1B p.S367 synonymous mutation, identified for the first time in our previous study (Demirci et al., 2021), in three patients and two controls. One of the patients with p.S367 (DAH19, 35 years old, female) had mild diabetes mellitus (DM), GDM, obesity, severe insulin resistance, hypertriglyceridemia, a measurable C-peptide level, and was not receiving any antihyperglycemic therapy. The other patient with p.S367 was a 42-year-old woman (DAH36). This patient had thyroid dysfunction, a measurable C-peptide level, and the absence of pancreatic islet autoantibodies.
However, since we observed this synonymous mutation in healthy controls, it was not considered an MODY-specific mutation in this study, and these two patients (DAH19 and DAH36) were diagnosed with T2DM based on their clinical/biochemical features. The other patient carrying p.S367 was a 41-year-old male diagnosed as HNF1B p.S362P/KLF11 P261L double heterozygosity, whose clinical features we described above (DAH62).
Homozygous mutations in
We detected a novel c.G411A (p.L137) synonymous mutation (located on the bHLH domain of the NeuroD1 protein) in the NEUROD1 gene in a 31-year-old female patient (DAH11). The patient had hypothyroidism and kidney cysts and menstrual irregularity. In this study, NEUROD1 p.T45A and p.P197H missense mutations were commonly detected in both patient and control, and, for that reason, not considered pathogenic. The p.P197H missense mutation in the NEUROD1 gene, which we previously detected in pediatric MODY patients (Demirci et al., 2021), was defined as a single-nucleotide polymorphism (SNP) in this study. This mutation involved in the neuronal helix-loop-helix transcription of the NeuroD1 protein was detected in six patients (DAH3, DAH5, DAH8, DAH10, DAH22, and DAH29) and 10 controls in our study group.
Two of these patients (DAH8 and DAH10) had neuropathy, one of the phenotypic features of MODY6. Five of the patients had no mutations other than p.P197H (DAH3, DAH5, DAH8, DAH10, and DAH29). One patient carrying the p.P197H mutation (DAH22) also carried the BLK mutation (p.A71T). The patient whose clinical phenotype was compatible with BLK-MODY was diagnosed as MODY11.
Mutations in the
In this study, we identified three missense mutations in the KLF11 gene (p.S182R, p.P261L, and p.R461Q). The male patient with the heterozygous KLF11 p.P261L mutation (DAH62) also carried the heterozygous HNF1B p.S362P missense mutation. The patient (female, 29 years old) who carried the KLF11 p.S182R mutation also carried the missense mutation PDX1 p.E69A (DAH33). The p.R461Q mutation, which was first identified in our previous study of pediatric MODY patients (Demirci et al., 2021), was found in a 43-year-old male patient (DAH60). This patient had high insulin requirements, hypercholesterolemia, neuropathy, nephropathy, and hypertension. The phenotypic effects of this mutation were observed to be more serious than in our previous study. The different effects of the mutation may be due to the longer duration of diabetes in adult patients.
Heterozygous mutations of the
The patient with deleterious p.I488T mutation was a 31-year-old female (DAH45) and she required insulin treatment, and had dyslipidemia, anemia, and obesity. The I488T mutation is localized in exon 10 of the CEL gene. This mutation was predicted to have a damaging effect on CEL protein by bioinformatic tools. The p.G157R and p.I336T mutations were found in the same patient (DAH44). The female patient who had insulin requirement, uncontrolled diabetes, elevated C-peptide, macrosomia, subclinical hyperthyroidism, retinopathy, neuropathy, stroke, and hypertension was also obese (BMI: 36.6 kg/m2). This patient also heterozygously carried the novel ABCC8 p.R1103Q mutation, which was detected as benign by bioinformatic tools.
The patient carrying the p.N493H mutation was a 39-year-old male (DAH23). The patient with low insulin requirement exhibited the clinical phenotype of moderately uncontrolled diabetes, mild obesity (BMI: 31.2 kg/m2), high C-peptide, hypercholesterolemia, high triglyceride and low HDL-C levels, retinopathy, nephropathy, and neuropathy.
We also observed two missense mutations (T412I and T93S) of the CEL gene with high frequency in both control and patient groups and evaluated them as SNPs. Of these, T412I was localized in the helical domain of the protein and was defined as “benign” in ClinVar. We found this SNP in 35 controls and 29 patients. To our knowledge, the T93S SNP was detected for the first time in this study. The T93S SNP located in the “heparin binding domain” of the protein was detected in eight controls and five patients.
There are rare
Since we detected p.L503P and p.A71T mutations in our control group as well as MODY patients in our study, we first thought that these mutations were not pathogenic for our society. However, Bonnefond et al. (2013) suggested that the A71T mutation may be mildly “diabetogenic” in the presence of obesity. The 71T mutant allele has been shown to reduce the half-life of the BLK protein (Delgado-Vega et al., 2012). When evaluated together with the report of Bonnefond et al., obesity (BMI: 32.5 kg/m2) and diabetes findings (FBG: 118 mg/dL and C-peptide: 3.1 ng/mL) of a 24-year-old female patient with A71T mutation (DAH22) were found to be compatible with the BLK-MODY phenotype. A71T mutation was also observed in one male (32 years old, BMI: 25.1 kg/m2, FBG: 80 mg/dL) and one female (36 years old, BMI: 22.1 kg/m2, FBG: 89 mg/dL) in the control group.
Our findings in the patient and control groups confirm that the A71T mutation has a diabetogenic effect in relation to obesity. On the contrary, the BLK p.L503P mutation was detected in one clinically diagnosed patient with MODY (DAH28) and one control subject. A 23-year-old male patient with p.L503P mutation was not obese (BMI: 19.1 kg/m2) and had mild diabetic symptoms (no insulin requirement and mild hyperglycemia). The control case, a 57-year-old female, had normal BMI (26.4 kg/m2) and FBG (99 mg/dL).
When we evaluated the results of all patients with BLK mutation together, we observed that the clinical effects of not only the A71T mutation but also other BLK mutations exacerbated in the presence of obesity. We also observed that hyperglycemic symptoms could be controlled with short-term (1–6 months) insulin therapy in nonobese patients with BLK mutation. In contrast, obese patients had uncontrolled diabetes requiring high-dose insulin (Supplementary Table S1).
Mutations in the
Previous studies showed that missense mutations in ABCC8 and KCNJ11 are very rare in the Turkish population (Arslan Ates et al., 2021; Demirci et al., 2021; Ozdemir et al., 2018; Yalcin Capan et al., 2020).
We identified known p.A1369S, p.V1572, and c.354C>T (p.V118V) and novel p.R1103Q, p.V421I, and p.K1411N mutations in the ABCC8 gene. Among these, the A1369S, V1572I, and K1411N were common in both MODY and control groups. The missense R1103Q and V421I and synonymous c.354C>T (p.V118V) mutations were MODY-specific. The missense mutations (p.R1103Q and p.V421I) presented in ClinVar, however, there are no studies in the literature on their phenotypic effects. Both mutations were located in the transmembrane region of the ABCC8 transporter protein. The p.R1103Q mutation was predicted to be benign, whereas p.V421I was predicted to be damaging.
The female patient with p.R1103Q mutation (DAH44) also carried missense CEL p.I336T (novel), CEL p.G157R (known), and novel synonymous KCNJ11 c.G561A (A157A) mutations. The patient had a phenotype compatible with MODY8/MODY12 double heterozygosity with the clinical features mentioned above in CEL mutations. The clinical features of a 36-year-old female patient (DAH12) carrying the V421I mutation had overweight, mild diabetes, and history of GDM and macrosomic baby. The V118V synonymous mutation was found in a 35-year-old female patient (DAH35). The patient's clinical features (mild obesity, uncontrolled diabetes, nephropathy, retinopathy, GDM, and hypertension) were consistent with the MODY12 phenotype.
To the best of our knowledge, this is the first report that associates these p.R1103Q and p.V421I mutations of the ABCC8 gene with MODY12.
Here, we identified p.L270V (Yalcin Capan et al., 2020) and p.K23E (Chistiakov et al., 2008; Pappa et al., 2011) SNPs in the KCNJ11 gene, which were previously associated with T2DM, in both patient and control groups. In the present study, the KCNJ11 p.S385F mutation, detected in the pediatric/adolescent MODY group in our previous study (Demirci et al., 2021), was observed in only two healthy controls. Therefore, it cannot be considered a pathogenic variant for the adult MODY group. In addition, the KCNJ11 K39E SNP was first observed in this study in both the patient and control groups (in MODY: n = 7; in controls: n = 4).
Conclusions
We have conducted a study in which genetic factors that may be the cause of MODY were evaluated together with the detailed anamnesis and clinical and laboratory parameters of the patients. This study, which analyzes the clinical effects of MODY-specific mutations in detail, draws attention to the importance of evaluating MODY mutations in comparison with a population-based control group. It is noteworthy that the causative MODY variations are different in different populations; however, some common variations are found in our study sample as well as in studies in many other populations.
The findings of our study support the genetic and clinical heterogeneity of MODY in our country, which is a transitional society with many subpopulations, and show the existence of new unique variations that cause MODY in our population. BLK (MODY11) gene mutations were found to be the most common cause of MODY in our study population as recruited from adult index patients and their diabetogenic effects were obesity-dependent. It has been observed that diabetic symptoms are more severe in the presence of obesity in patients with BLK mutation, and require long-term insulin therapy, while hyperglycemic symptoms can be controlled with short-term (1–6 months) insulin support in nonobese patients.
In addition, when we evaluated the MODY patients in our sample, along with their MODY-specific mutations, complications, and comorbidities, we observed that obesity, dyslipidemia, and other comorbid conditions in MODY patients may affect hyperglycemic symptoms and diabetes complications, and thus, treatment approaches and outcomes as in T2DM patients (Supplementary Table S1). A fact that further challenges the MODY in clinical practices is that it is already heterogeneous. Consequently, by revealing the MODY-specific mutations, NGS is currently the most reliable tool to inform the MODY clinic, which is intertwined with the symptoms/complications of other forms of diabetes, and thus, NGS offers an important tool to prevent misdiagnoses.
In addition, it has been observed that having double heterozygous mutations in MODY-related genes is associated with clinical aggressiveness of the disease, which may also contribute to MODY heterogeneity. Our study indicates that some of the MODY cases have defects in unknown MODY genes, referred to as “MODY X.” Therefore, attempts to identify novel causative MODY gene variants using whole-exome sequencing are required in future studies to elucidate the genetic profile of MODY in our population.
Several limitations of the study are noteworthy of emphasis here. First, the current findings of this study only involved a single center, and thus, it may not be appropriate to generalize our results to the entire population. Second, the MODY-associated APLL1 gene (MODY14) was not analyzed.
It is of critical importance to compare the results of NGS-based genetic analysis of MODY patients with healthy individuals from population studies, as well as to evaluate them together with bioinformatic and detailed clinical findings. Thus, it will be possible to predict the clinical course of MODY and to provide the most appropriate treatment to the patients.
In conclusion, NGS analyses of the adult patients with suspected MODY appear to be informative in a clinical context. Nevertheless, these findings warrant further clinical diagnostic research and development in other world populations suffering from diabetes with genetic underpinnings.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.