
Abstract
Death-associated protein kinase 3 (DAPK3) is a serine/threonine protein kinase that regulates apoptosis, autophagy, transcription, and actin cytoskeleton reorganization. DAPK3 induces morphological alterations in apoptosis when overexpressed, and it is considered a potential drug target in antihypertensive and anticancer drug development. In this article, we report new findings from a structure-guided virtual screening for discovery of phytochemicals that could modulate the elevated expression of DAPK3, and with an eye to anticancer drug discovery. We used the Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT), a curated database, as part of the methodology. The potential initial hits were identified based on their physicochemical properties and binding affinity toward DAPK3. Subsequently, various filters for drug likeness followed by interaction analysis and molecular dynamics (MD) simulations for 100 nsec were performed to explore the conformational sampling and stability of DAPK3 with the candidate molecules. Notably, the data from all-atom MD simulations and principal component analysis suggested that DAPK3 forms stable complexes with ketanserin and rotenone. In conclusion, this study supports the idea that ketanserin and rotenone bind to DAPK3, and show stability, which can be further explored as promising scaffolds in drug development and therapeutics innovation in clinical contexts such as hypertension and various types of cancer.
Introduction
Death-associated protein kinase 3 (DAPK3) is a calcium/calmodulin-regulated Ser/Thr kinase that regulates apoptosis, autophagy, transcription, and reorganization of actin cytoskeleton (Chuang et al., 2008). Depending on the cellular setting, it regulates smooth muscle contraction and both apoptotic and autophagic-dependent cell death signals (Usui et al., 2014). The condensation of actin stress fibers into dense bundles is also regulated by the overexpression of DAPK3, which is elaborated in actin filament focal adhesion dynamics (Das et al., 2016). It has enhanced transcription from androgen-responsive promoters in a hormone and kinase-dependent mode and regulates cell cycle progression and proliferation (Leister et al., 2008).
DAPK3 is involved in many human malignancies, such as hypertension (Carlson et al., 2018), smooth muscle disorders (Haystead, 2005), and various cancers (Elbadawy et al., 2018; Pike et al., 2008). DAPK3, in combination with Pim kinases, regulates contractility and controls hypertension (Carlson et al., 2018). A dual DAPK3/Pim inhibitor prescribed as HS56 and selective DAPK3 inhibitors, HS94 and HS148 have been developed to combat hypertension (Carlson et al., 2018). In another study, an automated molecular de novo design led to the discovery of a novel inhibitor of DAPK3 (Rodrigues et al., 2015).
Studies have shown that DAPK3 promotes cell proliferation in many cancers (Leister et al., 2008). It has been found to promote cell proliferation in prostate cancer (Leister et al., 2008). Also, the DAPK3 knockdown was found to prevent cell proliferation in colon cancer through the Wnt/β-catenin signals inhibition (Togi et al., 2011). Another study revealed that the DAPK3 knockdown also blocked non-small cell lung cancer progression through cellular signaling (Kake et al., 2017). This crucial piece of evidence suggests that DAPK3 is a reliable therapeutic target for hypertension, smooth muscle disorders, and different types of cancer.
DAPK3 structure has an N-terminal kinase domain, a leucine zipper domain, and two putative nuclear localization sequences (Elbadawy et al., 2018). The protein kinase domain is present at amino acid positions 13–275, whereas the activation segment region is present at positions 161–204. The leucine-zipper region is present on amino acid positions 427–441. The structure of DAPK3 provides a platform to develop novel small molecules targeting its activity (Rodrigues et al., 2015). We have collected a total of ∼9000 phytoconstituents from the Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT) database and filtered them by applying various structural parameters to narrow down our investigation to the most promising therapeutic candidates that target DAPK3.
IMPPAT is one of the largest manually curated databases constructed via literature mining from specialized books on traditional Indian medicine, published research articles, and other existing database resources. It contains >1700 Indian medicinal plants, >9500 phytochemicals, >1100 therapeutic uses, and >900 traditional Indian medicinal formulations. We evaluated the conformational flexibility and dynamic stability of DAPK3 in the presence of the elucidated phytoconstituents by utilizing all-atom molecular dynamics (MD) simulation for 100 nsec, principal component analysis (PCA), and free energy landscape (FEL).
Materials and Methods
Molecular docking-based virtual screening
The virtual screening of the compounds that exhibit high-affinity binding toward the protein of interest, DAPK3, was done with the help of state-of-the-art tools such as InstaDock (Mohammad et al., 2021), PyMOL (DeLano, 2002), and DS Visualizer (Studio, 2008). These tools helped analyze the interaction between the ligand molecules and their respective sites on the target protein (Anjum et al., 2021a, 2021b). To perform molecular docking, the refined structure of the protein was extracted from the Protein Data Bank (PDB) (Rose et al., 2017).
The protein crystal structure PDB ID: 3BHY was then subjected to preparation for virtual screening that is carried out with the help of SwissPDB Viewer (Guex and Peitsch, 1997). The tools help in re-modeling the protein by incorporating compatible residues in missing atoms, adding hydrogen atoms on the respective polar groups in DAPK3 to confer rigidity and specificity to intermolecular interactions, etc.
The retrieval of the phytochemicals or the potential compounds was done from the IMPPAT database enriched with plant derivatives with pharmacological activity (Mohanraj et al., 2018). The IMPPAT database provides a substantial source of extracting pharmaceutical compounds that can be filtered and investigated to extract highly selective therapeutics with minimal side effects. The compounds are researched and after subjecting them through various selective parameters, the compounds were selected to filter out the most promising compounds.
The compounds with good affinity scores were processed in InstaDock for interaction analysis. The close polar contacts established between the DAPK3 protein and the respective compounds were measured in the acceptable range of 3.5Å as close interactions in protein visualization software, PyMOL. Along with PyMOL, DS Visualizer was also used to identify possible interactions between the target protein and drug compounds. Subsequently, the molecules that interacted with the crucial residues in the protein's binding pocket were finalized for further analyses.
Physicochemical properties of compounds
During drug selection and development, the safety and efficacy of the drug play a decisive role. To examine the compound's efficacy for therapeutic purposes, the absorption, digestion, metabolism, excretion, and toxicity (ADMET) analysis helps to provide information on the physiochemical properties of the small molecule. ADMET analysis was accomplished through SwissADME (Daina et al., 2017) and pkCSM (Pires et al., 2015), where the files are prepared in Simplified Molecular Input Line Entry System (SMILES) strings and following the analysis and selecting the filtered results; Pan-assay interference compounds (PAINS) analysis was done to rule out the molecules with a tendency to interact with multiple sites on the target protein (Baell, 2016).
The criteria for the PAINS filter is taken as zero to avoid the selection of compounds that can result in side effects by binding to non-target proteins in the body. Such compounds have the tendency to bind to the non-desired targets and alter the healthy functioning physiology of the body. Therefore, a thorough and careful exploration of these properties of the selected molecules is vital for drug discovery and selection.
Prediction of activity spectra for substances analysis: biological activity predictions
Prediction of activity spectra for substances (PASS) analysis is done to evaluate the biological and pharmacological attributes of the compounds screened through the ADMET and PAINS filter (Lagunin et al., 2000). PASS or prediction of the activity of spectra for substance investigates the relationship between the structure and function of the compound and issues the biological potential to the molecule. The results are given in a ratio between the “probability of being active,” denoted as Pa, and “probability of being inactive,” denoted as Pi. The higher the value of Pa greater will be the biological property of the molecule that is under examination. This implies better bioavailability and drug-like properties of a compound.
MD simulations
The MD simulations were done on the HP Z480 system using the GROMACS simulation suite (Van Der Spoel et al., 2005). The GROMOS force field aided the simulation of DAPK3 in apo and ligand-bound conditions with ketanserin and rotenone. The PRODRG web server generated the force-field parameters and topology for ketanserin and rotenone (Schüttelkopf and Van Aalten, 2004). The steepest-descent algorithm employed energy minimization, and the position refinement was done through the number N of particles, volume V, and total energy E (NVT) and number N of particles, pressure P, and total energy E (NPT) ensemble.
The provision of water molecules was done through the SPC216 model, and only requisite counterions were introduced to balance the charge neutrality. Finally, the 100 nsec simulation was executed for each system, and the consequential trajectory was assessed through GROMACs' inbuilt tools. QtGrace software was used for simulation data visualization and plot generation (Turner, 2005).
PCA and FEL
The PCA approach is employed to understand proteins' folding mechanism and basic motions (Maisuradze et al., 2009). This provides crucial information of the functional structural aspects of a protein in the free state as well as when bound with the investigating ligands (Naqvi et al., 2018). The PCA is a mathematical method based on a covariance matrix and narrows down the multidimensional set of variables. The PCA studies and FEL give simulated trajectory and embody the diagonalization of eigenvectors to discover the conformational sampling of DAPK3 and the complex it forms with ketanserin and rotenone (Altis et al., 2008). The FELs impart stability analysis of DAPK3and its complexes with ketanserin and rotenone.
Results and Discussion
Molecular docking
The parent library of phytochemicals from the IMPPAT database was filtered through Lipinski's rule of five (RO5) to extract the compounds with drug-like properties (Lipinski, 2004). After applying RO5, a filtered library of 6093 compounds was retrieved from the IMPPAT database and subjected to the molecular docking process. Lipinski's RO5 accepts the compounds that have a molecular mass of <500 Daltons, <5 hydrogen bond (H-bonds) donors, <10 H-bonds acceptors, and the log p value of octanol-water partition to be <5 (Lipinski 2004). This process acted as filtering criteria to rule out compounds with lesser efficacy and binding potential toward DAPK3. The top 10 hits with substantial affinities with DAPK3 were fetched out with the calculated docking affinity score of −8.3 to −9.3 kcal/mol (Table 1).
Selected Hits and Their Docking Scores with Death-Associated Protein Kinase 3
Predicted inhibitory constant.
Ligand efficiency values are in kcal/mol/non-H atom.
ADMET properties of compounds
The ADMET analysis enables the screening of the compounds based on their absorption, digestion, metabolism, excretion, and toxicity (Daina et al., 2017). These attributes are important for analyzing a potential pharmacological compound to enhance clinical success. The pkCSM web server allows the ADMET analysis, the 10 compounds that exhibit the best-accepted attributes are filtered, and the results are displayed in Table 2. Only two compounds, that is, ketanserin and rotenone that do not show any toxic pattern and have good ADME activity were selected for further evaluation.
Absorption, Digestion, Metabolism, Excretion, and Toxicity Properties of the Selected Compounds
BBB, blood brain barrier; CNS, central nervous system; GI, gastrointestinal.
Ketanserin is a well-known antagonist of serotonin receptor hydroxytryptamine (5HT2) and works to reduce hypertension (Vanhoutte et al., 1988). Besides acting on the 5HT2 receptor, it also directly exerts its influence on serotonin receptor 2a and 2c (Hattori et al., 2017). Ketanserin along with naftopidil is shown to enhance the potentiating effect of alpha-methyl-serotonin on the neurally induced contraction of urinary bladder muscle strips (Hattori et al., 2017).
Ketanserin has also been observed to exhibit a suppressing effect on the alpha-Me-5 hydroxytryptamine (α-Me-5-HT), thereby acting on the serotonin system and reducing hypertension (Pettersson et al., 1985). Ketanserin in higher concentrations also inhibits the functioning of alpha-1-adrenoceptors, thereby positively regulating blood pressure (Hedner and Persson, 1988). Ketanserin is a potent vasodilator, regulating the dilation of capacitance vessels and resistance. It has also contributed to reducing cardiovascular morbidity and death rates in people with chronic hypertension (Hedner and Persson, 1988; Vikenes et al., 1999).
Rotenone is shown to inhibit the complex I of the electron transport chain in mitochondria, which can be explored to act on vital processes of cancerous cells (Deng et al., 2010; Lee et al., 2011). The presence of reactive oxygen species (ROS) as a result of oxidative stress can induce apoptosis in breast and lung cancer cells (Hu et al., 2016). The underlying mechanism for rotenone-dependent apoptosis still remains unexplored. The activation of NOX2 by rotenone, however, leads to the production of ROS that increases autophagy in cancerous cells (Hu et al., 2016).
PASS analysis: biological activity predictions
The PASS analysis includes screening relationships between the structure and activity. The training set of the PASS server provides the biological activities of chemical molecules based on their molecular features (Lagunin et al., 2000). After this prediction, ketanserin and rotenone were selected as they demonstrated anticancer activity and kinase inhibitory potential (Table 3). The compounds used as the reference provided similar results, indicating the potential of the selected drug constituents. The pa value of >0.7 supports the findings that compounds ketanserin and rotenone are suited candidates as antineoplastic, protein inhibitors, and tp53 expression enhancers. The elucidated molecules exhibit promising pharmaceutical properties that can be investigated further to develop selectively targeting anticancer therapies.
Selected Hits and Their Biological Properties Predicted Through Prediction of Activity Spectra for Substances Webserver
Pa, probability to be active; Pi, probability to be inactive.
Interaction analysis
After shortlisting the compounds through rigorous screening, the binding conformations of the docked compounds were exposed to interaction analysis with DAPK3. Simultaneously, the reference binding sites (Lys42, Asp139, Glu94, and Val96) were subjected to pose selection, and the obtained docked conformers were found to be superimposed with the protein crystal structure. This enables us to carry the investigation further. During analysis, the selected compounds ketanserin and rotenone formed interactions with the crucial residues of the binding site, including the active site Asp139 of DAPK4.
The interaction between the elucidated compounds and DAPK3 is exhibited in Figure 1. The interaction analysis shows that ketanserin and rotenone interact with residues in the binding pocket of DAPK3 and make different close associations with binding site residues (Fig. 1B). Both compounds fit inside the deep cavity of the DAPK3 binding site and block its action through virtuous complementarity, as shown in Figure 1C. Lys42, the adenosine triphosphate (ATP)-binding site of DAPK3, also participates in the interaction, which may prevent ATP accessibility to the protein and so render the protein ineffective in driving the tumorigenic pathways. The ATP provides the driving force for biochemical pathways and also serves as a potent target site for anticancer therapies.
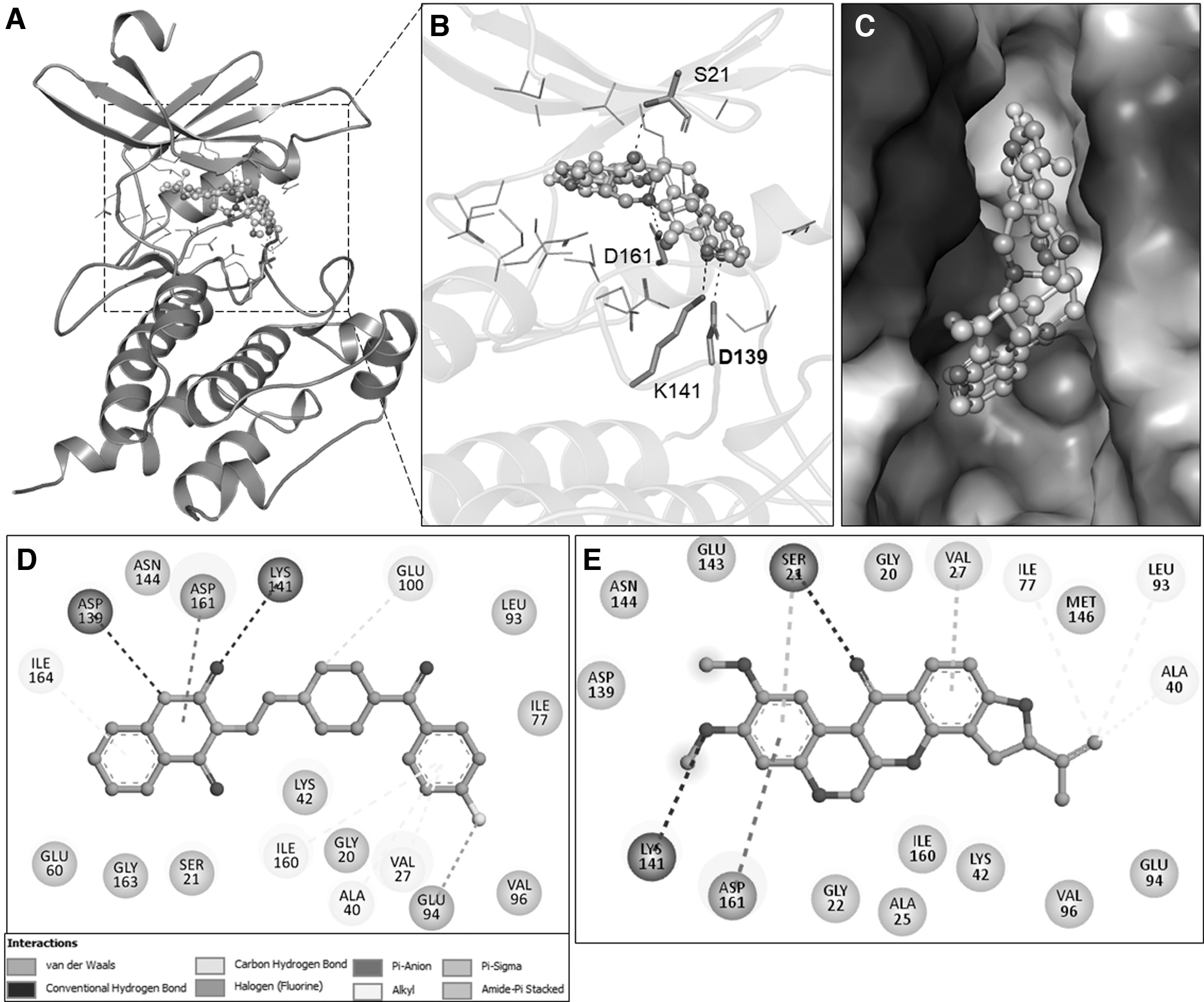
Organizational representation of DAPK3 interaction with ketanserin and rotenone.
The elucidated compounds ketanserin and rotenone accompany each other, facilitating various types of noncovalent interactions. Two-dimensional plots of ketanserin and rotenone expose their interaction between the crucial residues, that is, Lys42, Asp139, Glu94, and Val96, to suppress the functionality of DAPK3. A detailed visual analysis of the interaction between the elucidated compounds and DAPK3 is shown in Figure 1D and E.
MD simulations
MD enables further refinement of docked structures by exploring the flexibility between the proteins and respective ligand molecules (Ali et al., 2019; Naqvi et al., 2018). It is useful in bridging small structural details through dynamic evaluation of proteins restricted in experimental approaches (Fatima et al., 2020; Naz et al., 2018). The MD simulations carried out in a solvent environment assess the structural stability and dynamics for time. The docking complexes containing DAPK3, ketanserin, and rotenone were used in MD simulations for 100 nsec in synchrony with the time evaluation of various structural and dynamic parameters.
Structural changes and compactness
Proteins are dynamic structures that deviate from their structure over time; the root mean square deviations (RMSDs) help to understand such deviations and aid in structure analysis (Khan et al., 2021). The DAPK3 protein backbone alterations were subjected to RMSD analysis, and the results of DAPK3, DAPK3-ketanserin, and DAPK3-rotenone are shown in Figure 2A by plotting the deviations through the simulation trajectory.
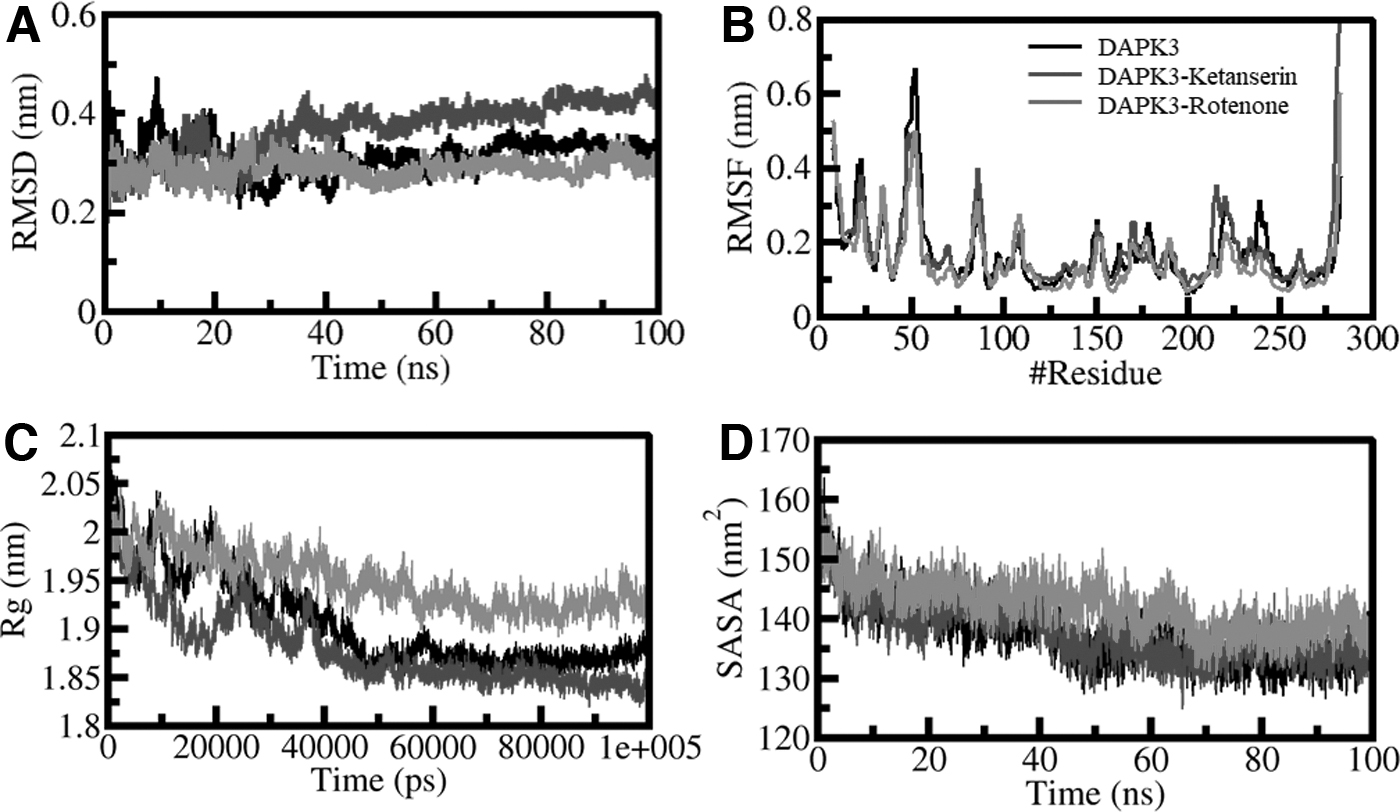
Structural dynamics of DAPK3 upon ketanserin and rotenone binding.
The RMSD graph showing small fluctuations in ligand-bound systems supports the long-term stability of complexes compared with unbound DAPK3. The three complexes achieved equilibrium and remained relatively stable during the simulation trajectory of 100 nsec, as explained in Figure 2A. The unprecedented deviations in the RMSD (∼0.1 nm) over 30 nsec are attributed to the system adjustment and comparing the DAPK3-ketanserin and DAPK3-rotenone complex. The trajectory later was to be more stabilized in the simulation period.
The RMSD graphed as a probability distribution function (PDF) illustrated the lower RMSD and stabilization in DAPK3 dynamics with a greater probability in the ligand-bound state of DAPK3. The details of the values are shown in Figure 2A. The analysis concluded that ligand-bound states of DAPK3 are stable throughout the simulation.
Root means square fluctuation (RMSF) provides information on the residual motions and vibrations present in the protein structure during the MD simulation (Mohammad et al., 2020a). The RMSF value of the DAPK3 and complexes DAPK3-ketanserin and DAPK3-rotenone in the protein backbone was evaluated to measure the flexibility of residues individually. The system that shows a similar pattern of RMSF is shown in Figure 2B. The graph plot illustrates that these fluctuations are dynamically stable and minimized with ketanserin and rotenone binding, supporting the ligand-protein interaction's stability studies.
The RMSF analysis provided information on the stability of the residues of DAPK3 while interacting with ketanserin and rotenone with only slight fluctuation. The residues that deviated higher than other systems stipulate the initial binding adjustments. An increase in the deviation in the DAPK3 after rotenone binding was restricted to the helical region. The MD simulation shows that the overall interaction between ketanserin and rotenone with DAPK3 is stable with time.
The radius of gyration (Rg) is an important tool that helps check the compactness of a protein (Mohammad et al., 2020b). The time of evolution of Rg was plotted to evaluate the compactness of DAPK3 in the ligand-bound and the apo states. The time evaluation of Rg was evaluated to check the DAPK3 compactness before and after ligand binding. Three systems (DAPK3-apo, DAPK3-ketanserin, and DAPK3-rotenone) were examined for their compactness during the simulation. The Rg plot advised that DAPK3 in the presence of ketanserin and rotenone becomes stable between 1.85 and 1.95 nm during the entire simulation (Fig. 2C). The comparative results revealed that the folding of DAPK3 was consistently stable after ketanserin and rotenone binding. The PDF plot also showed that Rg values are similarly distributed for DAPK3 before and after compounds binding.
The compactness parameters such as solvent-accessible surface area (SASA) signify the surface area available to the solvent for access and are crucial for estimating a protein's stability and folding pattern (Elbadawy et al., 2018; Richmond, 1984). The native contacts of the proteins and the SASA value reflect information on how the protein folds in a given environment. The time evaluation study on SASA was carried out on DAPK3 before and after binding with drug candidates ketanserin and rotenone. The plot did not show much deflection in the SASA values during the simulation.
This suggests that the structure of DAPK3 is considerably stable even when it binds to ketanserin and rotenone. The following is shown in Figure 2D. After correlation with Rg values, the SASA distribution pattern exhibits equilibration in all systems without hindering the folding process and compactness of the structure. The SASA pattern of the target protein DAPK3 with ketanserin and rotenone is described in Figure 2D.
Dynamics of H-bonds
Hydrophobic interactions and intramolecular H-bonds primarily stabilize the protein's structure (Jairajpuri et al., 2020; Khan et al., 2021; Mohammad et al., 2020a). The intramolecular H-bonds facilitate the folding and conformation of the protein. Therefore, the investigation of these bonds helps to examine the compactness in a protein and the conformation deflections it expresses. The dynamic molecular trajectories estimate the formation of H-bonds in DAPK3 in a time-evolution manner.
The results helped examine the dependability of the intramolecular hydrogen bonding before and after the protein binds to ketanserin and rotenone. The fluctuations in the number of H-bonds formed in DAPK3 in the unbound and bound (with ketanserin and rotenone) state are depicted in Figure 3. The plot constructed reveals that H-bonds formed were stable and responsible for establishing the geometry of protein structure. The presence of an increase in the H-bonds confirms that the complexes DAPK3-ketanserin and DAPK3-rotenone are more compact than individual proteins. The PDF analysis on H-bonds showing steadfastness during simulation is cited in Figure 3B.
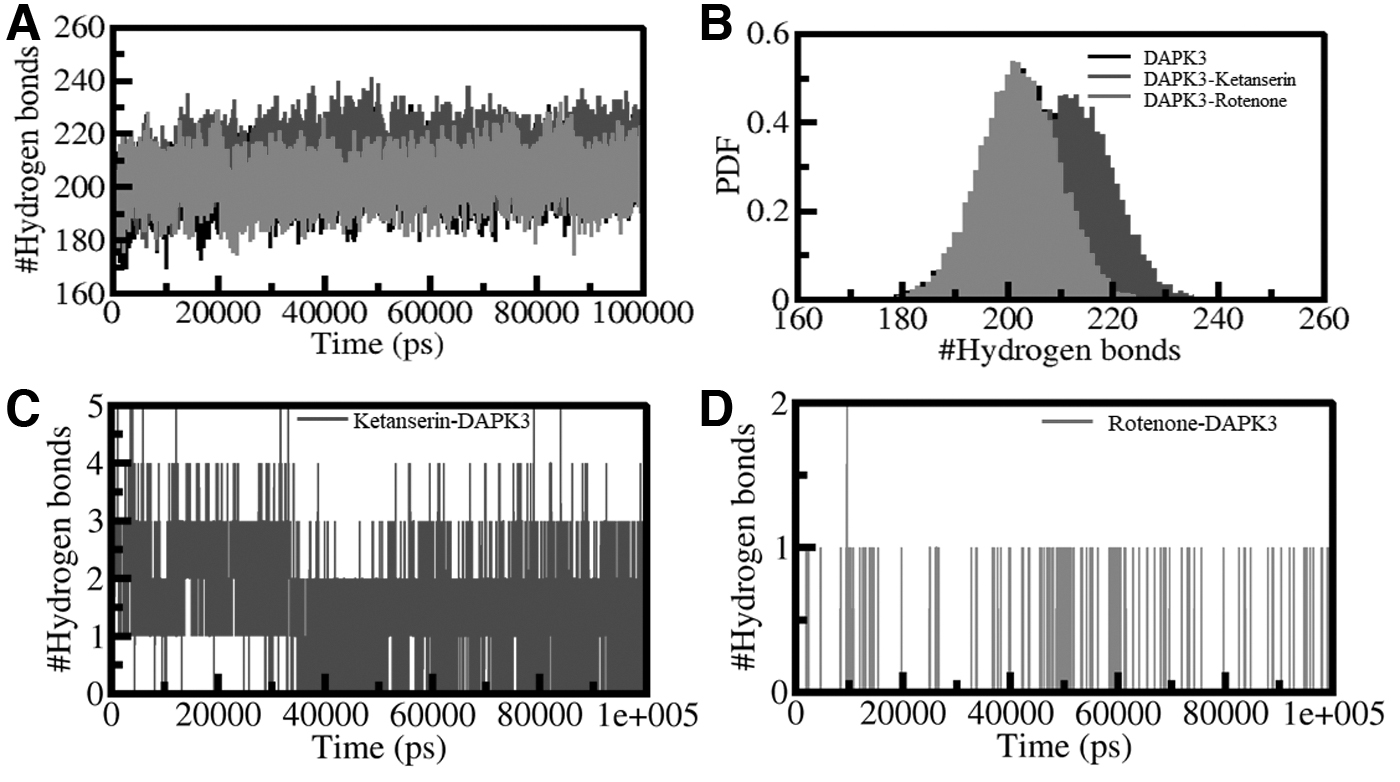
Intramolecular H-bonds.
The time-based evaluation assisted in exploring the reliability of the H-bonding of DAPK3 with ketanserin and rotenone. Inside the complex, the average number of bonds formed are two and one in the protein-ligand complexes, as shown in Figure 3. The H-bonds and higher PDF value indicate fair persistence in the H-bonding of the two systems. The molecules ketanserin and rotenone remained fixated on the initial docking position present on the target protein, concluding the complexes' relative stabilization.
PCA and FEL
The PCA is a useful approach to studying the impact of ligand binding on the collective motions of a protein receptor. The PCA was performed to inspect the conformational sampling of DAPK3-ketanserin, DAPK3-rotenone, and DAPK3 through the intervention of the essential dynamics approach (Maisuradze et al., 2009). The PCA helps to understand the target protein's collective movements and conformational sampling using simulated trajectories (Jairajpuri et al., 2020; Khan et al., 2021; Mohammad et al., 2020b).
Figure 4 shows the conformational sampling projected on the alpha carbon atoms present in the three systems in the essential subspace. The plotted data revealed that the DAPK3-ketanserin and DAPK3-rotenone complexes elementally occupied that same subspace as free state DAPK3. The free-DAPK3 shares the subspace of DAPK3-ketanserin in both the eigenvectors (EVs). The smaller flexibility subspace on EV1 encompassed by the DAPK3-rotenone complex decreases the flexibility of the DAPK3-ketanserin complex on EV. This supports the stability of the complex at the time of MD simulations.
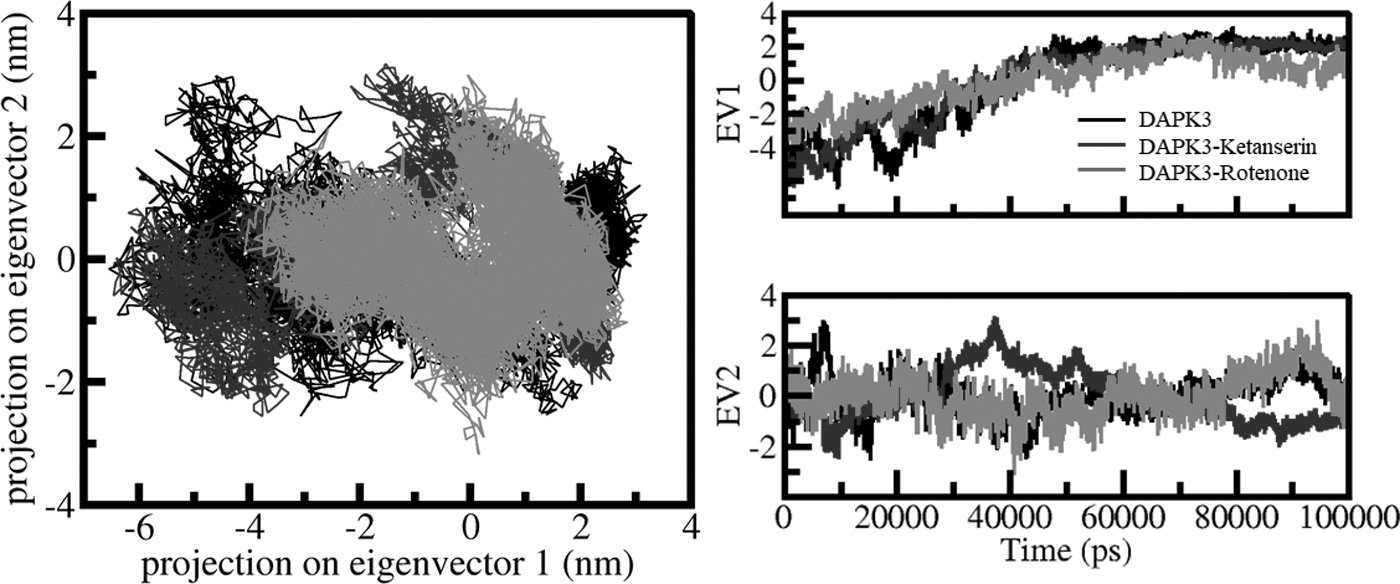
PCA. 2D projection representing the conformational sampling of DAPK3, DAPK3-ketanserin, and DAPK3-rotenone (left panel). The time evolution of DAPK3 projections on both EVs (right panel). EV; PCA, principal component analysis.
The FELs are utilized to understand the stability of the protein molecules and the complexes formed by proteins and ligands under solvent conditions (Altis et al., 2008). The FEL is employed to explain the mechanism of protein folding in native states and their denaturation. The energy minima and conformational landscapes of the three systems DAPK3, DAPK3-ketanserin, and DAPK3-rotenone are illustrated in Figure 5. Based on this analysis, the ketanserin and rotenone binding with the target protein slightly affects the position and the size of the phases present within the one to two stable global minima.
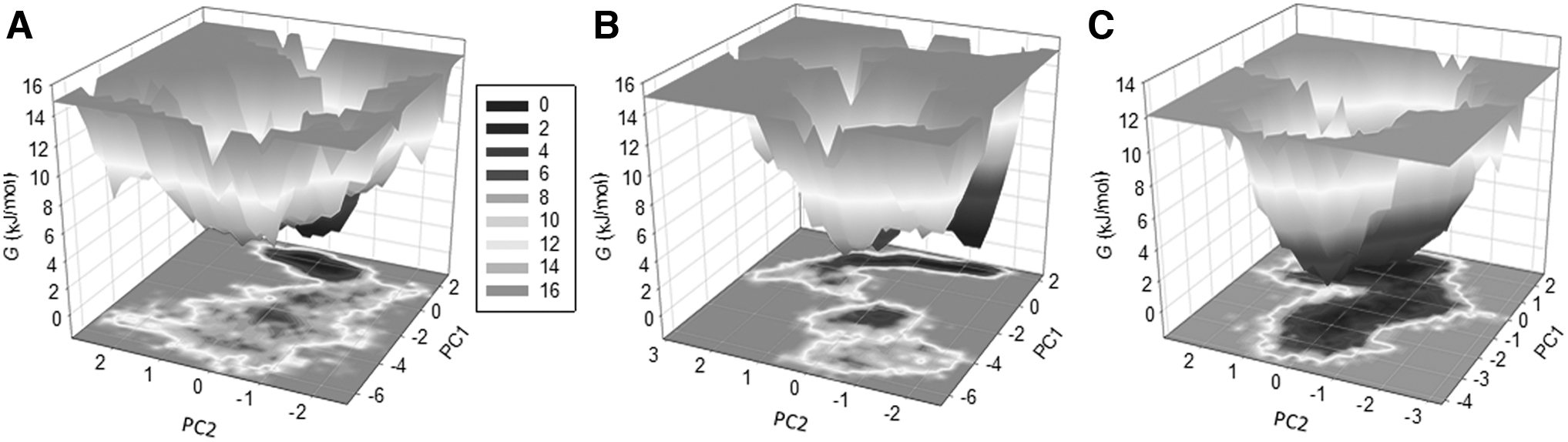
The FEL plots of
The darker shade in Figure 5 specifies the conformation with lower energy in their native states. The 3-dimensional plot also concludes that DAPK3 is restricted within the larger single global minima expanded to two to three basins. Similarly, the DAPK3-ketanserin and the DAPK3-rotenone obtain the smaller global minima with about two to three local basins with diverse populations. Overall, the dynamics and the simulation studies of the DAPK3 complex with the potential drug candidates ketanserin and rotenone recommend stability with minimum conformational shift during 100 nsec simulations.
Conclusions
Complex diseases such as hypertension and cancer have become widespread ailment that takes a toll on millions of lives each year. The constant struggle and investigation for the development of efficient treatment methods provide aspiration for modern therapeutics. The pharmacological suppression of the target protein DAPK3 is a promising approach for potential anticancer therapies. The study carried out aids in developing the therapeutic management of the diseases with the help of naturally available compounds.
DAPK3 is linked with positive regulation of cancer progression and hypertension, and the investigated compounds ketanserin and rotenone selectively target and inhibit DAPK3 to prevent its expression and thus can serve in potential anticancer and antihypertensive therapies. The in silico analysis is performed through established computational techniques. The identified compounds ketanserin and rotenone were filtered after conducting a rigorous exploration of the physiochemical and pharmacological properties of the phytoconstituents followed by their binding potential with DAPK3.
Hence, we put forth the two compounds ketanserin and rotenone for further research in vitro and in vivo for future anticancer and antihypertensive drug development and therapeutics innovation.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
A.M.E. extends his appreciation to the Deanship of Scientific Research at Jouf University for funding his work through Research grant no. (DSR-2021-01-0370). M.I.H. acknowledges the Council of Scientific and Industrial Research for financial support (project no. 27(0368)/20/EMR-II).