
Abstract
The hippocampus demonstrates age-associated changes in functions, neuronal circuitry, and plasticity during various developmental stages. On the contrary, there is a significant knowledge gap on age-associated proteomic alterations in the hippocampus subfields. Using tandem mass tag-based high-resolution mass spectrometry and quantitative proteomics, we report here age-associated changes in the human hippocampus at the subregional level. We used formalin-fixed paraffin-embedded hippocampal tissue sections from a total of 12 healthy individuals, with 3 individuals from each of the 4 different age groups, specifically, 1–10, 21–30, 31–40, and 81–90 years. We found that lysosome and oxidative phosphorylation were the pathways enriched in the 81- to 90-year age group. On the contrray, nervous system development, synaptic plasticity and transmission, messenger RNA (mRNA) splicing, and electron transport chain (ETC) complex-I activity were the enriched biological processes observed in the younger age groups. In a hippocampus subfield context, our topline findings on age-associated proteome changes include altered expression of proteins associated with adult neurogenesis with age in the dentate gyrus and increased expression of immune response-associated proteins with age in certain cornu ammonis sectors of the hippocampus. Signal peptide analysis predicted hippocampal proteins with secretory potential. While these new findings warrant replication in larger study samples, the current data contribute to (1) our understanding of the molecular basis of proteomic changes across various age groups in hippocampus subfields in healthy individuals, and (2) the design and interpretation of future research on the age-associated neurodegenerative disorders.
Introduction
Hippocampus is one of the most extensively studied brain structures. Hippocampus is highly vulnerable to various physiological factors, particularly age, which results in impairment of its basic functions, plasticity, and neuronal circuitries (Geinisman et al., 1995; Verret et al., 2007). Impairment in hippocampus cognitive functions at various developmental stages has been reported previously in mice, rat, monkey, and human (Gower and Lamberty, 1993; Lomidze et al., 2021; Rapp and Gallagher, 1996; Rosenzweig and Barnes, 2003).
Apart from that, studies have shown an increase in hippocampal atrophy with age in the cognitively normal populations (Golomb et al., 1993), at a rate of 10% per decade (Anderson et al., 1983). Several multiomics analyses of the whole hippocampus at different age groups (AG) have been reported, including proteomics of the human hippocampus during aging (Xu et al., 2016), age-related metabolomic profiling of the rat hippocampus (Durani et al., 2017), age-related changes in the proteomic profiling of rat (Freeman et al., 2009; Hamezah et al., 2018), and monkey hippocampus (Meng et al., 2020).
The hippocampus comprises a complex circuit with functionally interconnected and molecularly distinct subfields, including the dentate gyrus (DG), the four cornu ammonis (CA) sectors: CA1, CA2, CA3, and CA4, and the subiculum (Small et al., 2011). The regional vulnerability within the hippocampus to different developmental stages has drawn researchers to examine its specific subfields rather than the hippocampus as a whole (Lister and Barnes, 2009; Pereira et al., 2014). Several studies focused on age-associated alterations in different regions of the hippocampus, for example, the hippocampal CA3 (Adams et al., 2008) and the rat hippocampal CA1 (Shi et al., 2005), age-dependent changes in the synaptosomal proteins of rat hippocampus (Sato et al., 2005), and spatial learning in the rat hippocampal CA1 genes (Burger et al., 2007).
We also elucidated the proteomic profile of the five adult human hippocampal subfields (CA1, CA2, CA3, CA4, and DG), which suggested functional heterogeneity (Mol P. et al., unpublished data). However, given the magnitude of the studies on the hippocampus, we aimed to follow this up with an investigation of proteomic alteration with different age groups in the subfields of the human hippocampus in the present study.
There remains a significant knowledge gap on age-associated proteomic alterations in human hippocampus subfields. To obtain a deeper insight into the effects of age on the proteome of the hippocampus, we sought to investigate in the present study, the age-associated alterations in protein expression across its subfields using tandem mass tag (TMT)-based high-resolution mass spectrometry and quantitative proteomics. The current study reports new insights on the age-associated changes in the hippocampus as determined by quantitative proteomics of its five subfields to decipher the systems-scale molecular basis of age-associated changes in hippocampal proteome.
Materials and Methods
Formalin-fixed paraffin-embedded sample collection
This study was approved by the Ethics Committee of the National Institute of Mental Health and Neurosciences (NIMHANS, Bangalore, India). Written consents were obtained from the close relatives of the deceased tissue donors. The study included formalin-fixed paraffin-embedded (FFPE) hippocampal tissue sections from a total of 12 histopathologically normal individuals (Supplementary Table S1). Despite challenges associated with FFPE samples such as formaldehyde-induced crosslinking and protein denaturation (Maes et al., 2013; Magdeldin and Yamamoto, 2012), pressure cycling technology (PCT)-based protein extraction and high-throughput mass spectrometry analysis circumvented this challenge.
Accordingly, the sections were obtained from 1 to 10 years (n = 3, AG1), 21 to 30 years (n = 3, AG2), 31 to 40 years (n = 3, AG3), and 81 to 90 years (n = 3, AG4). Owing to well-known challenges in obtaining human brain samples, this age continuum for the study sample size could not include samples from several other age groups (41- to 79-year age group). However, we were able to cover the ages from early (1–10 years) and late (81–90 years) in the course of the human life span. Our current effort was in a modest total sample size of 12 cases (total n = 12), with 3 cases (n = 3) each from each different age group, as that of a draft map of the human proteome (Kim et al., 2014).
The FFPE sections (10 μm thickness) of the posterior hippocampus were collected from the Human Brain Tissue Repository (HBTR), NIMHANS. FFPE preservation of autopsy samples was performed using the standard set of procedures at the HBTR, NIMHANS.
Manual microdissection of hippocampal subfields
The hippocampal subfields were dissected from the FFPE sections after performing deparaffinization using a sequence of organic solvents such as xylene (5 min twice), 100% alcohol (2 min), and 70% alcohol (2 min). The tissue was further rehydrated with distilled water for 1 min. Manual dissection of subfields was performed under a stereo-zoom microscope (Olympus SZ81 with Q-Imaging Scientific Camera) using a microtip cataract knife. The neuropathology experts at NIMHANS (S.K.S. and A.M.) supervised the manual microdissection. The dissected tissues were collected in lysis buffer (4% sodium dodecyl sulfate [SDS], 50 mM triethylammonium bicarbonate [TEABC], and 100 mM dithiothreitol [DTT]). Figure 1A provides the images of hippocampal subfields captured during the manual microdissection.

Schematic illustration of experimental workflow. (A
PCT-based protein extraction
Cell lysis of the FFPE samples was carried out by the PCT, using a barocycler (Pressure Biosciences, Inc.) with 72 cycles of alternating pressure (40,000 psi for 50 sec and 5000 psi for 10 sec, which comprises 1 cycle) at 95°C. The samples were centrifuged at 12,000 revolutions per minute (rpm) for 15 min and the supernatant was collected. The extracted proteins were subjected to reduction using 10 mM DTT (30 min, 60°C), followed by alkylation using 20 mM iodoacetamide (IAA, 10 min in dark, room temperature). The proteins were then precipitated using chilled acetone (1:5) and subsequently resuspended in 50 mM TEABC buffer. Figure 1B shows the schematic illustration of the proteomic workflow used for this study.
Trypsin digestion and TMT labeling
The protein samples were digested using modified sequencing grade trypsin (1:50 ratio, 37°C, overnight; Promega, Madison, WI, USA) and labeled using 10-plex TMT reagents as per the manufacturer's instructions (Thermo Fisher Scientific) (Supplementary Table S1). Briefly, peptide digests were reconstituted in 50 mM TEABC (pH 8.5) and mixed with the corresponding TMT reagents dissolved in anhydrous acetonitrile and incubated at room temperature for an hour. Labeled peptides were normalized across the four age groups from each subfield based on the reporter ion intensities. Furthermore, the reaction was quenched by the addition of 5% hydroxylamine. For each subfield, the labeled peptides from the four age groups were pooled, vacuum dried, and stored at −80°C until fractionation.
Fractionation using basic reverse-phase liquid chromatography
The labeled peptides were fractionated using basic reverse-phase liquid chromatography (bRPLC) fractionation as described earlier (Selvan et al., 2014). Briefly, the labeled and pooled peptides of each subfield were reconstituted in bRPLC Solvent-A (1 mL, 5 mM ammonium formate, in 2% acetonitrile, pH 10). Peptides were resolved and fractionated with an increasing gradient of 3–95% bRPLC Solvent-B (5 mM ammonium formate, in 90% acetonitrile, pH 10) in XBridge C18, 5 μm, 250 × 4.6 mm column (Waters Corporation, Milford, MA, USA) in Agilent 1200 series high-performance liquid chromatography (HPLC) system with a flow rate of 500 μL/min for 120 min. The eluting peptides were collected in a 96-well plate and were concatenated to 12 fractions. The vacuum-dried samples were then subjected to C18-based StageTip clean-up before mass spectrometry analysis.
Mass spectrometry analysis
The liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of fractionated samples was carried out using the Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, Bremen, Germany) interfaced with the Proxeon Easy-nLC II system (Thermo Scientific) in technical triplicates. Briefly, samples were resuspended in 0.1% formic acid (liquid chromatography-mass spectrometry [LC-MS] grade) and loaded on a preanalytical column (2 cm × 75 μm, Magic C18 Aq; Michrom Bioresources, Inc.) with a flow rate of 3 μL/min. The resolution of peptides was carried out on an analytical column (20 cm × 75 μm, Magic C18 Aq) using an increasing gradient of 5% to 30% of solvent B (95% acetonitrile, 0.1% formic acid) at a flow rate of 300 nL/min for 120 min.
The mass spectrometer was operated in a data-dependent acquisition mode. The scan range was set to 350–1600 m/z using Orbitrap mass analyzer with a mass resolution of 120,000 at 200 m/z at MS level, and 50,000 resolutions at MS/MS level. Higher energy collision dissociation was selected as the fragmentation mode with 34% normalized collision energy. The automatic gain control (AGC) was set to 4 × 106 ions for full MS and 1 × 106 ions for MS/MS. Internal calibration was executed using lock-mass from ambient air (m/z 445.1200025).
Analysis of mass spectrometry data
The Proteome Discoverer (Version 2.2) software suite (Thermo Scientific) was used for protein identification and quantification. The LC-MS/MS data were queried against the Human RefSeq protein database (Version 89) using Mascot (Version 2.2.0) and Sequest HT search algorithms. The search parameters used were as follows: oxidation of methionine as a dynamic modification, carbamidomethylation of cysteine, TMT modification at N-termini of the peptide, and ɛ amine of a lysine residue as fixed modifications. Trypsin was specified as a protease with a minimum peptide length of 7 allowing two missed cleavages. Fragment ion mass tolerance and precursor ion mass tolerance were set to 0.05 Da and 10 ppm, respectively. The quantitation node in PD 2.2 was used to calculate the protein abundance ratios across the age groups of each subfield. The reporter ion intensities from unique peptides were considered for the calculation of protein abundances.
The false discovery rate (FDR) was calculated using a decoy database, and a cutoff of 1% FDR at peptide spectral matches (PSM), peptide, and protein levels was used for the identification.
Bioinformatics and Gene Ontology analysis
To calculate the abundance ratio, we used the protein abundance of the AG4 group as a control group and compared the protein abundance of the other three age groups against that for each subfield. Multiple t-tests were performed using Perseus software (Version 1.6.15.0) on the technical replicates across the age groups to find the statistically significant proteins of p-value ≤0.05 (Tyanova et al., 2016). Significantly altered proteins were identified by applying a fold change cutoff of 1.5 and a p-value ≤0.05. Gene Ontology (GO) analysis was carried out using the DAVID (Huang et al., 2009) functional annotation tool. The protein–protein interaction was performed using the STRING functional protein association network tool (Version 9.1) (Franceschini et al., 2013). N-terminal signal peptides for proteins were mapped using SignalP version 6.0 (https://services.healthtech.dtu.dk/service.php?SignalP) (Teufel et al., 2022).
Data availability
Mass spectrometry data generated in this study have been deposited to the Proteome Xchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE (Vizcaino et al., 2013) partner repository and are accessible using the data set identifier PXD031806.
Results and Discussion
Age-associated proteomic profiling of human hippocampus
The age-associated variations in the proteome of the human hippocampus were elucidated by comparing its proteomic alterations across the four different age groups specifically, 1–10 years (AG1), 21–30 years (AG2), 31–40 years (AG3), and 81–90 years (AG4). To get a deeper insight into the hippocampus, we choose to conduct the proteomic analysis across these four age groups at the subfield level, which resulted in the identification of 5698, 5508, 5428, 4271, and 4773 proteins in DG, CA4, CA3, CA2, and CA1, respectively (Table 1 and Supplementary Table S1). We report a significant trend in the protein expression pattern with age in the hippocampus. The total proteins identified across the age groups in each subfield are provided in Supplementary Table S2.
Summary of Total Proteins Identified, and Differentially Altered Proteins (at 1.5-Fold Cutoff) Identified in Each Age Group on Comparison with the Other Three Age Groups in This Study
AG, age group; CA, cornu ammonis; DG, dentate gyrus.
The Venn diagram provided in Supplementary Figure S1 shows the common and subfield-restricted proteins identified in the five hippocampal subfields. We found 3520 proteins common to all subfields that include growth-associated protein-43 (GAP43), brain-abundant membrane attached signal protein-1 (BASP1), and synapsin-1 (SYN1). Whereas 571, 268, 253, 46, and 100 proteins were found to be restricted to DG, CA4, CA3, CA2, and CA1, respectively. Calsequestrin-1 (CASQ1, p = 7.75 × 10−09) and calsequestrin-2 (CASQ2, p = 4.55 × 10−06) were examples of DG restricted proteins. Interleukin-1 receptor accessory protein-like-1 (IL1RAPL1, p = 5.02 × 10−06), prolactin receptor isoform-5 (PRLR, p = 8.00 × 10−04), elafin (PI3, p = 4.47 × 10−09), and transcription factor Sp5 (SP5, p = 5.15 × 10−06) were examples of proteins restricted to CA1, CA2, CA3, and CA4, respectively.
The older (81–90 years) age group displayed a distinct proteomic profile compared with other groups
The hierarchical clustering of statistically significant proteins (p ≤ 0.05) across the age groups showed a distinct protein expression profile in each subfield as depicted in the heat maps (Fig. 2A–E) and the principal component analysis (PCA) plots (Fig. 2F–J). Interestingly, clusters of proteins—Cluster 1 and Cluster 2, were observed to be of higher and lower abundance, respectively, in AG4 compared with the other three age groups irrespective of subfields. As major alterations in protein expression were significantly observed in the AG4 compared with the younger age groups, our major focus was on the functional analysis of proteins altered in AG4 compared with the other three age groups.
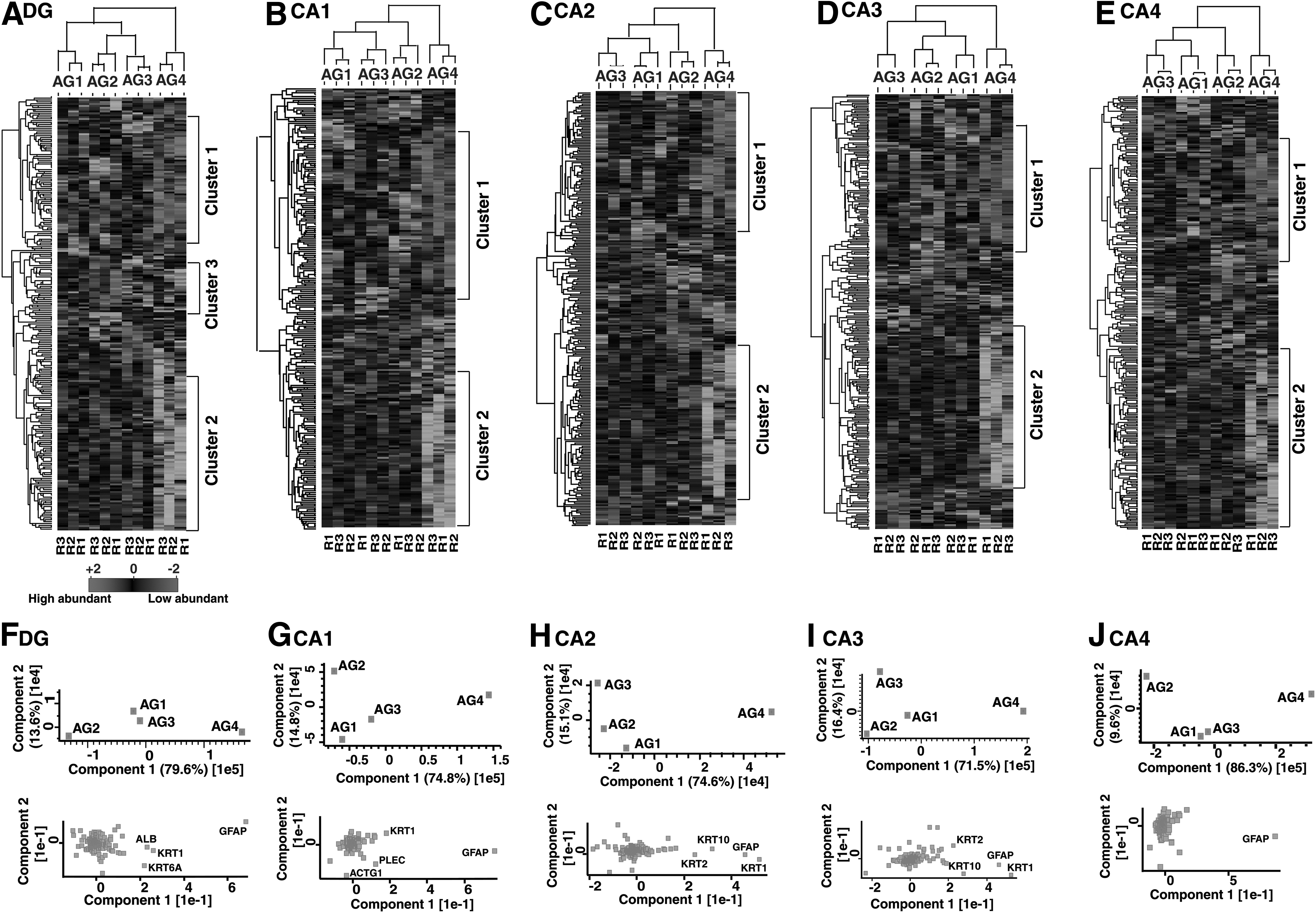
(
The GO enrichment analysis of protein clusters that were altered in the AG4 of each hippocampal subfield was carried out, and the most enriched biological processes are discussed below. A partial list of proteins associated with the biological processes found enriched in these clusters is provided in Supplementary Table S3.
Proteome-level alterations across age groups in the DG
The higher abundance proteins in AG4 of DG (Cluster 1 of Figs. 2A and 3A) were mostly enriched in platelet degranulation. Platelet activation and consequently degranulation processes have been reported to increase in the elderly, the age when infections and inflammations are more prevalent (Hearps et al., 2012; Montenont et al., 2019). However, the lower abundance proteins in the AG4 compared with the other age groups in DG (Cluster 2 of Figs. 2A and 3A) highlighted biological processes such as assembly of mitochondrial electron transportcomplex-1, G1/S transition of the mitotic cell cycle, and neuron projection morphogenesis. The role of these processes in hippocampal neurogenesis, granule neuron maturation, and regulation of cell proliferation has been reported earlier (Bertoli et al., 2013; Emmerzaal et al., 2020; Wu et al., 2008).

(
These findings suggest the role of proteins associated with these processes in cell differentiation and postnatal neurogenesis in AG1 to AG3 in DG. Studies reported a gradual decline in adult neurogenesis with age resulting in compromised hippocampal functions (Kempermann et al., 2002; Kuhn et al., 1996).
Proteome-level alterations across age groups in CA1
The homeostatic process and cellular oxidant detoxification were the enriched biological processes associated with the higher abundance proteins in AG4 compared with the other age groups in CA1 (Cluster 1 of Figs. 2B and 3B). As age progresses, brain regions are vulnerable to mechanisms, such as free radical generation, inflammation, organelle dysfunctions, and a series of insults and injuries, that severely affect processes serving cognition and plasticity in the hippocampus (Zia et al., 2021).
Cellular oxidant detoxification is a crucial homeostatic process that removes toxic free radicals and prevents oxidative stress from causing injury to neurons (He et al., 2017). Whereas proteins that are of higher abundance in AG1, AG2, and AG3 in CA1 compared with the AG4 (Cluster 2 of Figs. 2B and 3B) in CA1 showed enrichment in the ephrin receptor signaling pathway, which is significant in the migration of neurons, axon guidance (Cramer and Miko, 2016), formation of dendritic spines, and synaptic plasticity in hippocampal CA1 neurons (Grunwald et al., 2004).
Proteome-level alterations across age groups in CA2 and CA3
Proteins that are found to be of higher abundance in the AG4 compared with the other age groups of CA2 (Cluster 1 of Figs. 2C and 3C) highlighted neutrophil degranulation and innate immune response. Neutrophils are the major effectors of cell innate immunity, and neutrophil degranulation is one way of attacking pathogens by releasing proinflammatory substances during inflammation in older age groups (Lacy, 2006).
Therefore, these two processes are important in the older age groups due to the increased incidence of infection and inflammation in the older ages. We also found keratinization and cornification to be enriched in the AG4 of both CA2 and CA3 subfields (Cluster 1 of Figs. 2C, D and 3C, D). These processes are associated with skin renewal and have been reported to slow down dramatically with age (Farage et al., 2013). Although the role of the hippocampus in the skin renewal process has not been reported in the literature, these findings necessitate further validation.
Higher abundance proteins in AG1, AG2, and AG3 compared with the AG4 of CA2 (Cluster 2 of Figs. 2C and 3C) were enriched in the microtubule and actin cytoskeleton organization, which is reported to be very crucial in maintaining the neuronal homeostasis (Guillaud et al., 2020), neuronal polarization, and neurotransmission (Kapitein and Hoogenraad, 2011).
The proteins of lower abundance in the AG4 compared with the other three age groups (Cluster 2 of Figs. 2D and 3D) were enriched in chemical synaptic transmission in CA3, which underlies the basis for learning and memory (Kobayashi et al., 1996). A balanced chemical synaptic transmission in the DG-CA3 circuit is important in determining the formation and/or updating of memory (Miller and Sahay, 2019).
Proteome-level alterations across the age groups in CA4
The proteins of higher abundance in the AG4 of CA4 compared with the other age groups (Cluster 1 of Figs. 2E and 3E) were most enriched in the oxidation/reduction process. Oxidation/reduction reactions in glycolysis and oxidative phosphorylation are the processes through which the brain transduces energy for various neuronal activities such as synaptic transmission. Alterations in the mitochondria energy-transducing capacity and neuronal glucose uptake are well-known hallmarks of the aged brain that can lead to age-associated cognitive impairment (Yin et al., 2016). However, proteins found to be of lower abundance in the AG4 of CA4 compared with the other age groups (Cluster 2 of Figs. 2E and 3E) were most enriched in the mitochondrial electron transport complex-1, which plays a central role in energy metabolism for various neuronal activities (Franco and Vargas, 2018).
Pathway enrichment analysis of altered proteins in the older age group
Pathway enrichment analysis of the altered proteins in AG4 (1.5 cutoff) compared with the other three age groups of hippocampal subfields revealed the lysosomal pathway as an enriched pathway markedly influenced in the older age group of all the five subfields. Oxidative phosphorylation was found to be another enriched pathway associated with the older age group (AG4) of all the subfields except CA3. Studies on cognitive status and normal aging in mice hippocampus, and rat hippocampus revealed oxidative phosphorylation as the top canonical pathway associated with the onset of memory deficit in the older brain (Hamezah et al., 2017; Neuner et al., 2017). A list of proteins associated with oxidative phosphorylation is provided in Supplementary Table S4.
Lysosome pathway and neuronal homeostasis
The lysosome pathway is the primary cellular pathway capable of degrading damaged organelles, lipids, and protein aggregates (Finkbeiner, 2020). It also maintains cellular homeostasis (Kobayashi and Kageyama, 2021; Peng et al., 2019), synaptic plasticity, and synthesis of myelin and neurotransmitter (Ashraf et al., 2018; Nikoletopoulou and Tavernarakis, 2012). Lysosomal functions such as autophagy and lysosomal acidification cease as age progresses affecting cellular homeostasis and mitophagy. Mitophagy dysfunction leads to elevated reactive oxygen species production—an underlying factor in the pathogenesis of age-related disorders (Finkbeiner, 2020; Wang et al., 2019).
However, the role of the lysosomal pathway in the older brain and/or hippocampus is not fully understood (Peng et al., 2019). Housekeeping lysosomal protein, prosaposin (PSAP), and lysosomal aspartyl protease, cathepsin-D (CTSD), were the examples of proteins found associated with this pathway and altered in AG4 (p ≤ 0.05) compared with the other age groups. A list of proteins associated with this pathway is provided in Table 2.
Proteins Associated with Lysosomal Pathway and Their Expression Across the Age Groups in Hippocampal Subfields
Abundance ratio given in italics and bold italics represents high and low abundance (1.5-fold cutoff) respectively. p-Values highlighted in bold are statistically significant (p ≤ 0.05).
Signal peptide analysis of hippocampal proteins across the age groups
We performed signal peptide analysis of hippocampal proteins and examined their age-associated proteomic expression in each subfield. We found 41, 12, 26, 41, and 43 signal peptide-containing proteins that were significantly altered (p ≤ 0.05, 1.5-fold cutoff) across age groups in DG, CA1, CA2, CA3, and CA4, respectively, that constitute <1% of total proteins identified in the corresponding hippocampal subfields (Supplementary Table S5). Cellular localization annotation revealed that the majority of these proteins are localized to extracellular space, endomembrane system, cytoplasmic vesicle, secretory granule, and lysosome.
Examples of proteins with secretory and/or transmembrane potential identified in this study include CASQ2 with a high probability signal peptide score (Sec/SP1) of 0.999 that was found to be sevenfold higher abundant (p = 4.6 × 10−06) in AG4 compared with other age groups in DG. Of note, CASQ2 was identified as a DG-restricted protein in this study. We found calreticulin (CALR, Sec/SP1 = 0.999) to be 1.7-fold lower abundant in AG4 (p = 1.70 × 10−08) compared with other age groups of CA2. Another example was elafin (PI3, Sec/SP1 = 0.999), which was identified as a CA3-restricted protein in this study and was sixfold higher abundant (p = 4.47 × 10−09) in AG4 compared with other age groups.
Hippocampus subfields and their association with neurological disorders
Hippocampus gets primarily affected in different orders and varying degrees in various age groups and neurological disorders with unknown etiology (Babcock et al., 2021; Gao et al., 2022). In this study, we examined the expression of proteins implicated in major neurological disorders such as Alzheimer's disease (AD) and major depressive disorder (MDD) across the age groups in human hippocampal subfields. A recent study reported 1071 genes differentially regulated in MDD in the microarray profile data set of mouse DG (Wei et al., 2021). We compared the current DG protein data set with their data set and found 185 genes common in both studies. The age-associated proteomic alterations of these MDD implicated proteins in DG and the AD implicated proteins reported in the literature (Brinkmalm et al., 2014; Davis et al., 2017; Liu et al., 2019; Ohrfelt et al., 2016) identified in this study are provided in Supplementary Table S6.
Conclusions
Age-associated changes in the proteome across a broad age range of the human hippocampus subregions identified both shared and distinct differences in protein expression across the age groups, and at the hippocampus subfield level. We found proteins associated with adult neurogenesis and central nervous system development to be more abundant in the younger age groups. The lysosomal pathway and oxidative phosphorylation were the enriched pathways in the older age group. These findings are consistent with previously reported literature while also breaking new ground and insights into age-associated proteomic alterations in five human hippocampal subfields. While these new findings warrant replication in larger study samples, the current data contribute to the following: (1) our understanding of the molecular basis of proteomic changes across various age groups in hippocampus subfields in healthy individuals, and (2) the design and interpretation of future research on the age-associated neurodegenerative disorders.
Author's Contributions
T.S.K.P., A.M., and S.K.S. conceptualized and designed the study. S.K.S. and A.M. provided FFPE sections for the study and supervised the manual microdissection of FFPE samples. P.M., O.C., and L.G. collected the samples. P.M., L.G., S.K.S., and T.S.K.P. optimized the dissection strategies of subfields of the hippocampus on FFPE sections. P.M. carried out the manual microdissection. P.M., L.G., and O.C. carried out the sample preparation and fractionation under the guidance of T.S.K.P. K.K.M. and F.B. carried out mass spectrometry analysis. P.M., L.G., O.C., and M.K. analyzed the mass spectrometry data. P.M. wrote the initial draft of the article. P.M. and O.C. prepared the figures and tables. T.S.K.P., A.M., and S.K.S. critically reviewed the article. All authors read and made a significant intellectual contribution to the final article contents, and approved the final version of the article.
Footnotes
Acknowledgments
The NIMHANS-IOB facility is supported by the DBT Program Support on Neuroproteomics and infrastructure for proteomic data analysis. We also thank the Human Brain Bank Repository, NIMHANS, for providing human brain samples for the study. We thank the Ministry of Health and Family Welfare, Government of India, for providing instrumentation to the NIMHANS-IOB laboratory, NIMHANS. P.M., L.G., and O.C. are recipients of INSPIRE Senior Research Fellowship from the Department of Science and Technology (DST), Government of India.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No specific funding was received for this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.