
Abstract
Kidney renal cell carcinoma (KIRC) is the most common type of renal cancer. Kidney malignancies have been ranked in the top 10 most frequently occurring cancers. KIRC is a prevalent malignancy with a poor prognosis. The disease has risen for the last 40 years, and robust biomarkers for KIRC are needed for precision/personalized medicine. In this bioinformatics study, we utilized genomic data of KIRC patients from The Cancer Genome Atlas for biomarker discovery. A total of 314 samples were used in this study. We identified many differentially expressed genes (DEGs) categorized as upregulated or downregulated. A protein-protein interaction network for the DEGs was then generated and analyzed using the Search Tool for the Retrieval of Interacting Genes plugin of Cytoscape. A set of 10 hub genes was selected based on the Maximum Clique Centrality score defined by the CytoHubba plugin. The elucidated set of genes, that is, CALCA, CRH, TH, CHAT, SLC18A3, FSHB, MYH6, CAV3, KCNA4, and GBX2, were then categorized as potential candidates to be explored as KIRC biomarkers. The survival analysis plots for each gene suggested that alterations in CHAT, CAV3, CRH, MYH6, SLC18A3, and FSHB resulted in decreased survival of KIRC patients. In all, the results suggest that genomic alterations in selected genes can be explored to inform biomarker discovery and for therapeutic predictions in KIRC.
Introduction
Renal cell carcinoma (RCC) is the most malignant tumor derived from the kidney. It has been on the rise for at least four decades. Kidney and renal pelvis malignant tumors have been placed in the top 10 most frequent cancer observed in the United States (Siegel et al., 2022). Kidney renal cell carcinoma (KIRC) is the most common subtype of renal cancer and has a high mortality rate (Hsieh et al., 2017) and a poor prognosis rate (Chen et al., 2022; Hu et al., 2019; Li et al., 2021a; Xu et al., 2021).
There are numerous risk factors for RCCs, including hypertension and obesity (Gray and Harris, 2019). Patients with stage one or two RCC have a 5-year survival rate of 80% to 90%. In the later stages, the rate drops to 71% and further down to 14% when the tumor becomes metastatic (National Cancer Institute, 2021). Metastasis may be observed in a large population of patients at diagnosis (Gupta et al., 2008). Studies suggest that more than half of the diagnosed patients with RCC are asymptomatic and are diagnosed incidentally. Therefore, a thorough comprehension of the mechanism of KIRC and the development of early diagnostic strategies are essential (Yin et al., 2020a; Yin et al., 2020b).
RNA-based molecular markers are often used as biomarkers (Habib et al., 2022; Xi et al., 2017). Many studies suggest that the expression of genes involved in the tumor might be related to the prognostic survival time of patients (Zhan et al., 2015). Therapeutic inhibition of overexpressed genes has been reported to significantly impact the carcinogenesis of several tumor types (Olivier et al., 2010). Inhibition of selected targets has been proven to be an effective method of inhibiting cancer.
In this study, we explored the genes involved in the onset and progression of KIRC. We obtained differentially expressed genes (DEGs) between the tumor and normal cells of KIRC from The Cancer Genome Atlas (TCGA). The study shows a comparative analysis of genes employing annotated genes, functional analysis, protein-protein interaction (PPI) analysis, and survival analysis. A selection of the most related genes as hub genes was generated, and many genes may play a significant role in the poor prognosis of KIRC. A graphical representation of the workflow used in this study is illustrated in Figure 1.
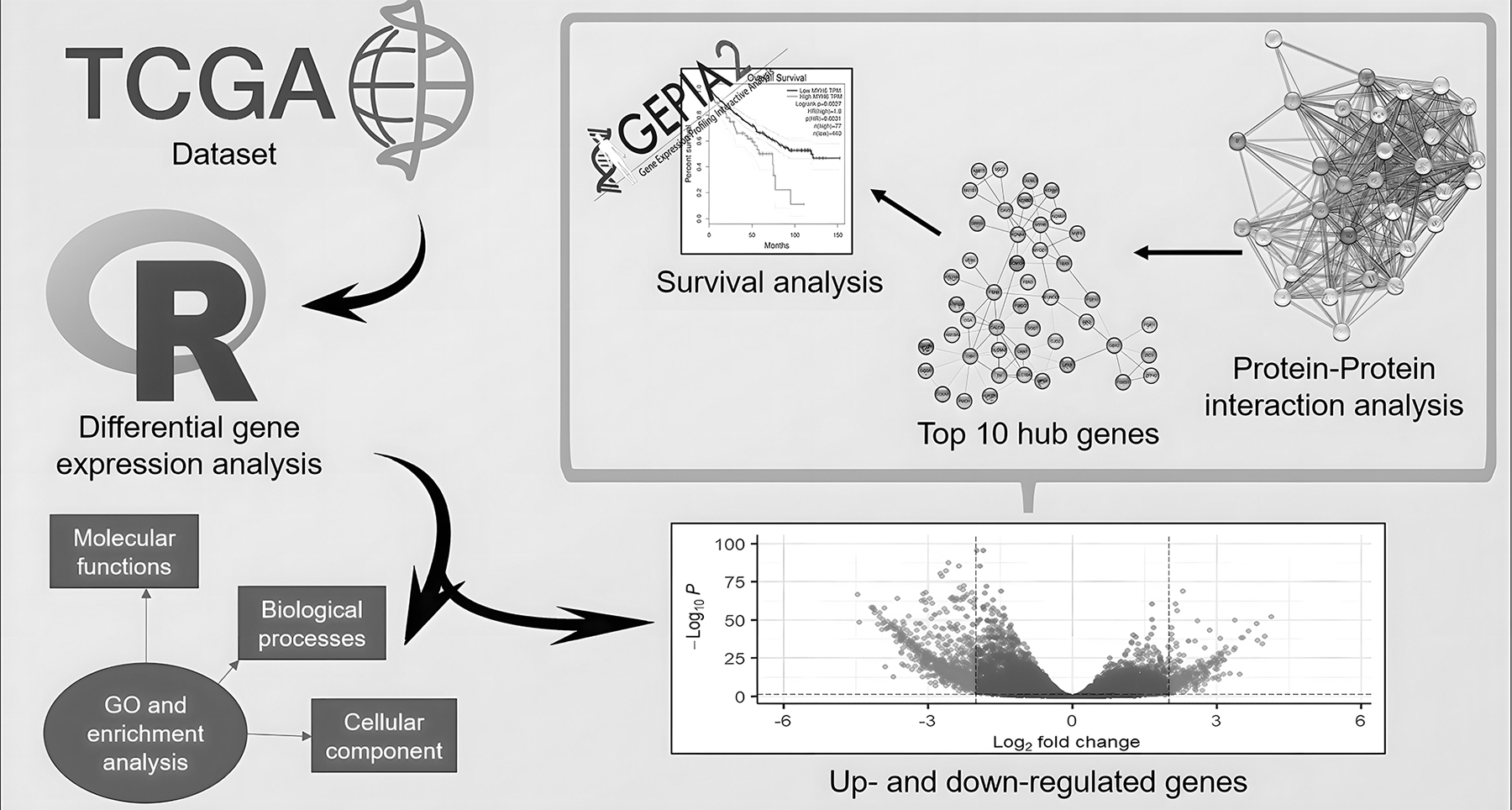
A graphical depiction of the workflow employed to study the effects of DEGs in KIRC. DEG, differentially expressed gene; HR, hazard ratio; KIRC, kidney renal cell carcinoma.
Materials and Methods
Data acquisition
The data for bioinformatics analyses were acquired from TCGA (https://portal.gdc.cancer.gov/). We employed the filter functions of the exploration tab of the browser to select the study of KIRC. The project TCGA-KIRC (Akin et al., 2016) was selected. The control and tumor KIRC samples were retrieved from the server. A total of 314 cases were obtained from the website. For each sample, an expression count for 60,484 genes was obtained from the server. This study used publically available data, and ethics committee approval and informed consent were not applicable.
Differential gene expression
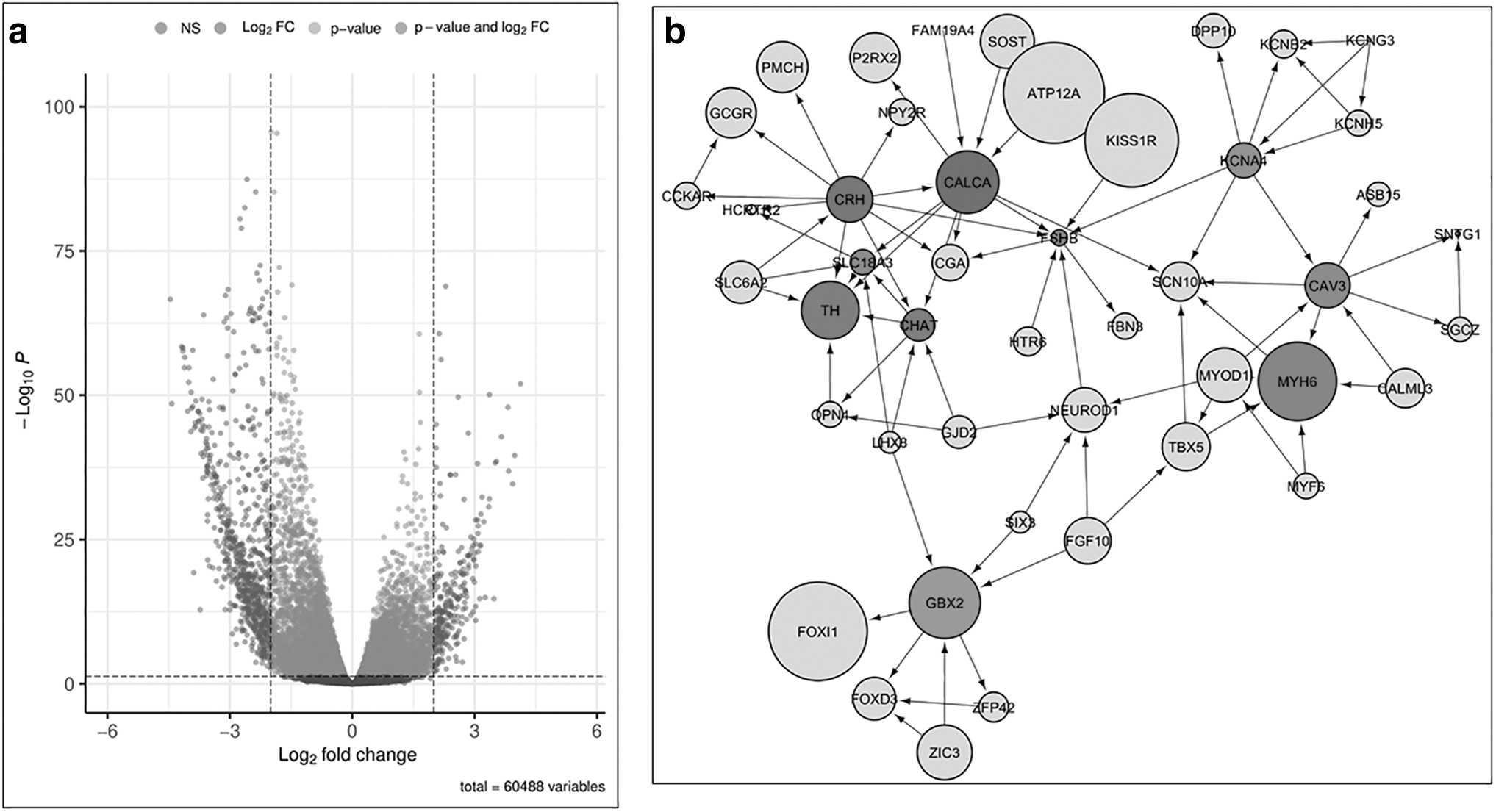
R, a scripting language (version: 4.2.1), was employed to perform differential expression analysis on the collected data. The DESeq2 library was used for differential gene expression analysis (Love et al., 2014). It required count data obtained from TCGA and creating a metadata file with samples and their respective tissue types. A log2fold change threshold of 2 and a p-value threshold of 0.005 were set in the R script. Using the “EnhancedVolcano” package (Blighe and Rana, 2022), a Volcano plot of log2fold change and p-value was generated (Fig. 2a). The “AnnotationDbi” package (Pagès et al., 2022) was then used to annotate and extract useful information for the obtained DEGs.
Functional analysis
Pathway and function analysis were necessary to understand the relatedness of the DEGs. For the functional analysis, the DAVID server (Sherman et al., 2022) was employed, which uses information from Gene Ontology (GO) (Mi et al., 2019) and KEGG (Kanehisa et al., 2021) databases. GO server outputs knowledge of the given genes' biological processes, cellular compartments, and molecular functions. The analysis provides the biological processes enriched in a group of genes.
PPI analysis
Search Tool for the Retrieval of Interacting Genes (STRING) server (Szklarczyk et al., 2019) integrated with Cytoscape (Shannon et al., 2003) was employed for identifying the PPIs. The interacting network was accessed using the UniProt IDs of annotated genes.
Hub gene analysis
CytoHubba add-in (Chin et al., 2014) of Cytoscape was used to obtain the top 10 hub genes with the highest scores and connectivity based on the Maximal Clique Centrality (MCC) method. The CytoHubba add-in is an appropriate method for ranking nodes in a network by their features. MCC is a precise predicting method explicitly devised for ranking essential genes or gene products.
Genetic alteration analysis
We have taken 2213 samples from 2137 patients under 12 different kidney cancer studies from TCGA. The elucidated genes CALCA, CRH, TH, CHAT, MYH6, SLC18A3, FSHB, CAV3, KCNA4, and GBX2 were chosen to explore in these samples. These genes were analyzed to generate an OncoPrint to analyze their genomic alterations in all the samples. In this study, different genetic alterations were profiled and compared with the previous studies. Alterations' frequencies were calculated for each genomic alteration in different cancerous populations.
Survival analysis
The top 10 genes were then analyzed for survival using Gene Expression Profiling Interactive Analysis 2 (GEPIA2) (Tang et al., 2019). We employed the survival analysis module of the server to construct the overall survival curves for hypothesizing the top key genes for the prognosis.
Results
DEG analysis
Using R, only genes that had a log2fold change and p-value greater than the threshold were retrieved and their expression was considered significant. From the values acquired, a negative fold change indicated downregulated genes, while a positive fold change indicated upregulated genes. The unannotated genes were removed from the dataset. After filtering, 309 downregulated genes and 77 upregulated genes were obtained from the analysis. Supplementary Tables S1 and S2 depict the top 25 upregulated and downregulated genes, respectively.
GO analysis
A total of 258 genes were found to be correctly mapped. The server provided functional annotations and enrichment analysis from GO and KEGG pathway databases. Thirty-four clusters were obtained from the functional annotation clustering method of DAVID. The largest cluster of genes was categorized as secretory cellular components in the biological transport process. Supplementary Table S3 depicts the top 5 clusters accessed from the DAVID server with their respective predicted significance and gene names. Supplementary Table S4 shows the KEGG pathways generated on the DAVID server. The KEGG pathways analysis indicated the role of elucidated genes in various critical pathways associated with various complex diseases, including cancer.
PPI network analysis
A total of 386 DEGs were submitted to Cytoscape for network analysis. The STRING plugin was used to generate the PPI network. A total of 138 nodes and 189 edges were obtained from the application, with an average number of neighbors being 2.739 and a clustering coefficient of 0.195. The resultant network was analyzed for kidney tissues to get results specific to KIRC. Topological parameters were analyzed as described in Supplementary Table S5 to explore the fundamental properties of the PPI network.
Hub gene analysis
Using the CytoHubba add-in of Cytoscape, we observed the most significant genes in the kidney tissue. The top 10 genes were ranked according to the MCC method based on the network-generated scores (Fig. 2b). The most significant hub genes were CALCA (Calcitonin-related polypeptide Alpha), CRH (Corticotropin-releasing hormone), TH (Tyrosine hydroxylase), CHAT (Choline O-acetyl transferase), MYH6 (Myosine heavy chain), SLC18A3 (Solute Carrier Family 18), FSHB (Follicle-Simulating Hormone B), CAV3 (Caveolin 3), KCNA4 (Potassium voltage-gated channel), and GBX2 (Gastrulation and brain-specific homeobox protein). Network Analyzer was used to calculate the topological parameters of the hub genes. Supplementary Table S4 depicts the summary statistics of the top 10 genes. The top 10 genes were ranked according to the MCC method based on the network-generated scores (Supplementary Fig. S1). CALCA, CRH, TH, CHAT, MYH6, SLC18A3, FSHB, CAV3, KCNA4, and GBX2 possess therapeutic potential in various domains.
Originally, CALCA showed promise in treating migraines and cardiovascular disorders (Alkanli et al., 2018). CRH, involved in the stress response, is targeted for depressive and anxiety disorders (Holsboer and Ising, 2008). TH, essential for neurotransmitter synthesis, is implicated in neurological and psychiatric conditions (Pearl et al., 2007). CHAT, responsible for acetylcholine production, is explored in Alzheimer's disease (Ferreira-Vieira et al., 2016). MYH6 mutations are associated with cardiac disorders (Razmara and Garshasbi, 2018). SLC18A3 dysfunction is linked to psychiatric disorders (Shinwari et al., 2017). FSHB plays a role in reproductive functions (Anderson et al., 1997).
CAV3 mutations affect muscle function (Catteruccia et al., 2009). KCNA4 mutations are tied to cardiac arrhythmias (Kang et al., 2023). GBX2's role in brain development has implications for neurodevelopmental disorders (Szulc et al., 2013). KEGG pathway analysis indicated their role in various critical pathways associated with complex diseases, including cancer. Hence, the study suggests that these genes might be associated with the development of KIRC. Understanding these genes and their products can inform targeted therapies. Further research is needed to unlock their full therapeutic potential.
Genetic alterations
The generated OncoPrint of samples shows different genomic alterations in the selected genes. In this study, overall, all the 10 genes (CALCA, CRH, TH, CHAT, MYH6, SLC18A3, FSHB, CAV3, KCNA4, and GBX2) were altered in 3% (a total of 72) samples out of a total of 2213 samples (Fig. 3). Maximum alterations were found in MYH6, which is 0.8%, mainly by amplification and deep deletion. At the same time, the other genes were altered with different frequencies. These alterations are accrued by amplification, deep deletion, missense, truncating, and fusion at many sites. At the same time, cancer type summary and alteration frequency show that the selected genes are altered up to 13.66% of 644 samples of kidney cancer (Supplementary Fig. S2).
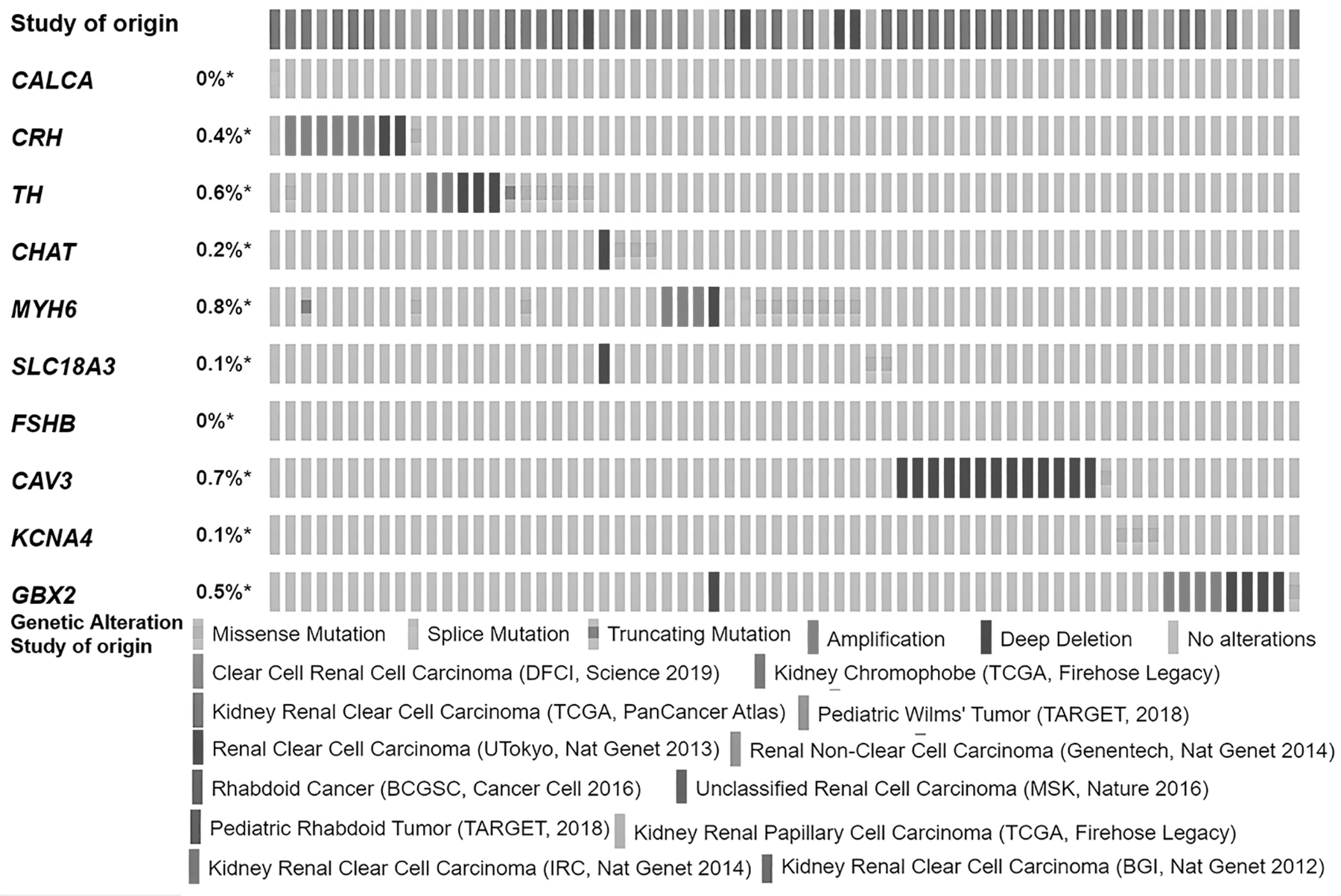
OncoPrint of genes and their genomic alterations in different populations of kidney cancer in TCGA. TCGA, The Cancer Genome Atlas.
Survival analysis
GEPIA2 was employed for the overall survival analysis of the top 10 selected hub genes. In the case of KCNA4, GBX2, TH, and CALCA, there was no significant difference between their expression levels. However, higher expression led to a lower overall survival rate in the case of CHAT, CAV3, CRH, and MYH6. The CRH expression level contributed to the most loss in survival rate. Genes FSHB and SLC18A3 also seem to affect survival. Figure 4 depicts patients' overall survival estimation plot for each of the top 10 selected hub genes.

Overall Kaplan-Meier survival estimation plot of KIRC patients for the top 10 CytoHubba selected genes form the TCGA dataset. A confidence interval of 95% was given for the Hazard Ratio (HR) used in every survival plot. The lighter lines denote increased expression while the darker lines denote lowered expression of the gene.
Discussion
Identifying biomarkers for KIRC is important due to its high prevalence and poor prognosis. This study aimed to analyze genomic data from KIRC patients obtained from TCGA to identify potential biomarkers and gain a deeper understanding of the disease. The study included 314 sample cases, and statistical and bioinformatics analyses were performed to investigate the expression patterns of genes in KIRC. The DEGs were identified and categorized as upregulated or downregulated, and a PPI network for the DEGs was generated and analyzed.
Using the MCC score defined by the CytoHubba plugin, a set of 10 hub genes was selected. These genes, namely, CHAT, CAV3, CRH, MYH6, SLC18A3, and FSHB, were observed to affect survival by overexpressing. An array of functions was applied to the initial data to obtain the results. The study discusses valuable information on KIRC and how the resulting genes can be targeted for further research, from expression analysis to PPI network analysis to survival rate analysis. Herein, we discussed the obtained 10 hub genes and their significance with respect to KIRC.
CALCA is a protein-coding gene that encodes calcitonin, a peptide hormone of significant impact. Diseases associated with CALCA include reflex sympathetic dystrophy and spinal stenosis. Among its related pathways are GPCR downstream signaling and the presynaptic function of kainite receptors (Rodriguez-Moreno and Sihra, 2004). GO annotations related to this gene include identical protein binding and protein-containing complex binding. The count data analysis hints at the upregulation of CALCA in KIRC. Calcium homeostasis is a crucial cellular function. A sustained calcium increase can affect cancer cell proliferation; studies suggest an increase in metastatic medullary thyroid carcinoma (Fan et al., 2022; Martinelli et al., 2017). Calcium levels can also affect endometrial cancer proliferation (Huang et al., 2022). Survival plot analysis inference suggests that higher expression of CALCA has a slight effect on the survival rate of cancer patients.
CRH gene belongs to the family of genes encoding Corticotropin-releasing hormone factors. The encoded protein, when processed, generates the mature neuropeptide hormone. It has been associated with colon cancer and inflammation in the gut (Baritaki et al., 2019). The study showed that expression levels of CRH were statistically significant, and higher values were linked to a lower risk of metastasis in colon cancer. However, for colitis-associated cancer models, CRH expression promoted colon cancer cell proliferation (Fang et al., 2017). The DEG analysis suggests that CRH is downregulated in KIRC. In the survival analysis, it can be observed that higher CRH led to a lower survival rate when compared to lower expression of CRH in patients.
TH is a gene encoding tyrosine hydroxylase, an enzyme that catalyzes the conversion of L-tyrosine to L-dihydroxyphenylalanine (Tolleson and Claassen, 2012). The DEG analysis suggests that TH is downregulated in KIRC. TH expression does not have a significant impact on the survival rate of patients.
CHAT is a gene encoding the neurotransmitter acetylcholine (Wilcock et al., 1982). No study suggests CHAT promotes any cancer cell growth. However, CHAT has been identified as a biomarker for Alzheimer's disease (DeKosky et al., 2002; Hampel et al., 2018). The DEG analysis suggests that CHAT is upregulated in KIRC. However, in the survival analysis, it can be observed that higher CHAT expression levels led to a lower survival rate when compared to lower expression of CHAT levels in patients.
SLC18A3 is a vesicular amine transporter family member. The protein encoded is a transmembrane transporter protein. The function of the encoded protein is to transport acetylcholine into the secretory vesicles to be released in the extracellular space. A study suggests SLC18A3 notably facilitated renal cancer cell proliferation through acetylcholine (Tie et al., 2022). A higher expression level of SLC18A3 was observed in tumor cells compared to normal nonmetastatic and noncancerous cells. The survival plot analysis carried out in this study converges to the same conclusion that high SLC expression does significantly impact patients' survival rate. It decreases the overall survival rate when compared to low SLC expression levels. The DEG analysis suggests that SLC18A3 is upregulated in KIRC.
FSHB is a pituitary glycoprotein hormone family member. The B or Beta part of the family protein is variable, and this specific gene encodes the beta subunit of Follicle-Stimulating Hormone (Bianco et al., 2021; Fan and Hendrickson, 2005; Nagirnaja et al., 2010; Trevisan et al., 2019). The hormone enables ovarian folliculogenesis to the antral follicle stage and is essential for Sertoli cell proliferation. Although the expression level of FSHB seems to impact patients' survival rates significantly, no existing study suggests that FSHB has a significant effect on the expansion of cancer cells. The DEG analysis suggests that FSHB is downregulated in KIRC.
MYH6 encodes the cardiac alpha-myosin heavy chain protein (Lu et al., 2022; Mahdavi et al., 1984). The heavy chain is an integral part of the Type 2 myosin protein. The protein helps generate the mechanical force required for the cardiac muscles to contract and relax. The survival plot suggests a decrease in the survival rate of patients with higher MYH6 expression levels. The DEG analysis suggests that MYH6 is downregulated in KIRC.
CAV3 is a gene that encodes the protein caveolin 3, the main component in caveolae, small pouches in the muscle cell membrane (Koga et al., 2003; Rothberg et al., 1992; Song et al., 1996). The survival plot suggests a decrease in the survival rate of patients with higher CAV3 expression levels. The DEG analysis suggests that CAV3 is downregulated in KIRC.
KCNA4 is the most complex class of voltage-gated ion channels from functional and structural standpoints. It has been associated with gliomas (Weng and Salazar, 2021) and gastric cancer (Zheng et al., 2011). The DEG analysis suggests that KCNA4 is downregulated in KIRC. KCNA4 expression level does not have a significant impact on the survival rate of patients.
GBX2 is a gene encoding a DNA-binding transcription factor. It has been associated with bladder cancer (Xiong et al., 2022), prostate cancer (Gao et al., 1998; Jeyapala et al., 2019), and gliomas (Li et al., 2021b). The DEG analysis suggests that GBX2 is upregulated in KIRC. GBX2 expression level does not have a significant impact on the survival rate of patients.
The OncoPrint results obtained from cBioPortal offer an insight into the genetic alterations occurring. The results provide inference by employing different populus of Kidney Renal Clear Cell Carcinoma in the TCGA database. Deep deletions and amplifications play a prominent role in the case of CRH, MYH6, CAV3, GBX2, and TH. Missense mutations could be observed in TH, MYH6, KCNA4, and SLC18A3. A total of 3% of the entire sample space of KIRC patients had genetic alterations. Genes such as FSHB and CALCA had very low to zero genetic alterations.
Conclusions
Our study collected RNA-Seq count data from the TCGA-KIRC project, which was then analyzed for gene expression. Initially, we identified a total of 386 DEGs. Out of the total DEGs, 77 upregulated and 309 downregulated genes were identified for the given data. Functional analysis of genes suggested that the highest number of genes clustered by the pathway analysis were of the signaling domain, followed by the ion transport process. Using the CytoHubba and STRING add-ins of Cytoscape, we selected the top 10 genes with the highest score according to the MCC method.
Using the GEPIA2 server, we generated survival rate plots for the top 10 genes and analyzed them for their impact on the overall and disease-free survival of KIRC. Six of the identified genes, CHAT, CAV3, CRH, MYH6, SLC18A3, and FSHB, were observed to affect survival by their differential expression and genomic mutations. Overall, this study suggests that the identified genes can be explored for future therapeutic and diagnostic contexts in KIRC after experimental validation, with a particular emphasis on SLC18A3 since it has been observed in renal cancer in previous studies.
Footnotes
Acknowledgments
The researchers would like to acknowledge Deanship of Scientific Research, Taif University for funding this work.
Authors' Contributions
Y.M.: Conceptualization, methodology, software, formal analysis, data curation, writing—original draft, and writing—review and editing. A.S.: Formal analysis, writing—original draft, and writing—review and editing. B.A.: Writing—original draft and writing—review and editing. Amal Adnan Ashour: Methodology, investigation, visualization, validation, writing—original draft, and writing—review and editing. W.A.A.-S.: Writing—review and editing. H.H.A.: Writing—review and editing. Salem Hussain Alharethi: Formal analysis, data curation, and review and editing. F.A.: Writing—review and editing, supervision, and project administration. All authors made a significant intellectual contribution, read, and approved the final article.
Data Availability Statement
The data underlying this article are available within the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The researchers would like to acknowledge Deanship of Scientific Research, Taif University, for funding this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.