
Abstract
Prostate cancer (PCa) represents a huge public health burden among men. Many susceptibility genetic factors for PCa still remain unknown. In this study, we performed a large splicing transcriptome-wide association study (spTWAS) using three modeling strategies to develop alternative splicing genetic prediction models for identifying novel susceptibility loci and splicing introns for PCa risk by assessing 79,194 cases and 61,112 controls of European ancestry in the PRACTICAL, CRUK, CAPS, BPC3, and PEGASUS consortia. We identified 120 splicing introns of 97 genes showing an association with PCa risk at false discovery rate (FDR)-corrected threshold (FDR <0.05). Of them, 33 genes were enriched in PCa-related diseases and function categories. Fine-mapping analysis suggested that 21 splicing introns of 19 genes were likely causally associated with PCa risk. Thirty-five splicing introns of 34 novel genes were identified to be related to PCa susceptibility for the first time, and 11 of the genes were enriched in a cancer-related network. Our study identified novel loci and splicing introns associated with PCa risk, which can improve our understanding of the etiology of this common malignancy.
Introduction
Prostate cancer (PCa) is a common malignancy and cause of mortality among men. With an estimated 1.6 million men diagnosed with PCa and 366,00 deaths attributed to it annually (Pernar et al., 2018), the growing and aging population is projected to result in almost 2.3 million new PCa cases and 740,000 related deaths annually by 2040 (Culp et al., 2020). Strong evidence from family studies supports a genetic predisposition to PCa (Eeles et al., 2008). For PCa, previous genome-wide association studies (GWASs) have identified 269 genetic risk variants, which explain <44% of its familial relative risk (Wu et al., 2019b). Transcriptome-wide association study (TWAS) integrating gene expression and genomic data sets has been applied to identify candidate disease susceptibility genes for multiple diseases, including PCa (Gusev et al., 2018; Mancuso et al., 2018; Wu et al., 2019b; Wu et al., 2018).
To date, previous TWASs have identified more than 800 candidate susceptibility genes for PCa risk (Emami et al., 2019; Fiorica et al., 2020; Gusev et al., 2019; He et al., 2022; Liu et al., 2022; Mancuso et al., 2018; Wu et al., 2019b). However, these loci and genes still fall short in fully explaining the genetic susceptibility of PCa, which calls for additional efforts to identify novel genetic factors for PCa.
As a posttranscriptional regulatory mechanism, alternative splicing through premessenger RNA (mRNA) molecules can produce many distinct mRNAs (Raj et al., 2018) that change the structure of transcripts and their encoded proteins (Stamm et al., 2005). This is the main source of protein diversity due to 90% of human gene alternative splicing expression (Martinez-Montiel et al., 2018). It has been reported that more than 15,000 alternative splicing events are associated with cancer biology (Marasco and Kornblihtt, 2023). Such alternative splicing events have become a distinguishing feature for cancer, with strong potential for developing effective prognostic and therapeutic options (Martinez-Montiel et al., 2018). Alternative splicing has also been implicated to play key roles in normal tissue development (Chen and Weiss, 2015), and aberrant splicing can promote the proliferation, growth, and survival of cancer cells (Bradley and Anczukow, 2023; Chen and Weiss, 2015).
Aberrant alternative splicing yielding protein isoforms can also affect cell phenotypes and survival of PCa patients (Rajan et al., 2009). The critical role of alternative splicing in PCa development has already been suggested in previous studies (Carstens et al., 1997; Gusev et al., 2019; Mancuso et al., 2018; Munkley et al., 2017; Paschalis et al., 2018). For example, androgen receptor (AR) transcription factor is a major driver of PCa pathology (Munkley et al., 2017), and AR splicing variants have been implicated in the development and progression of PCa (Martinez-Montiel et al., 2018). Fibroblast growth factor receptor 2 (FGF-R2) splicing is shown to be associated with androgen insensitivity in human prostate tumors and loss of FGF-R2 isoform b can lead to progression of human PCa (Carstens et al., 1997; Paschalis et al., 2018).
However, a comprehensive understanding of splicing events across the genome related to PCa risk is largely lacking. There have been five conducted TWASs for PCa risk that identified more than 800 candidate susceptibility genes for PCa (Emami et al., 2019; He et al., 2022; Liu et al., 2022; Mancuso et al., 2018; Wu et al., 2019b). However, these studies largely focused on assessing the overall gene expression but not splicing expression. Although splicing expression was also studied in the study of Mancuso et al. (2018), only prostate tumor-derived splicing expression genetic prediction models are used. It is known that for studying PCa susceptibility, normal tissue is more accurate due to that the splicing expression could have been changed during tumor development for many introns (Liu et al., 2020).
Herein, leveraging a large available reference data set (the Genotype-Tissue Expression, GTEx) for normal prostate tissue, we performed a comprehensive splicing TWAS (spTWAS) using complementary alternative splicing expression genetic prediction model building methods elastic net (ENET) (Zou and Hastie, 2005), least absolute shrinkage and selection operator (LASSO) (Baselmans et al., 2019), and minimax concave penalty (MCP) (Zhang, 2010), to identify splicing introns for PCa risk.
Materials and Methods
Building splicing intron expression genetic prediction models
Three sets of splicing intron expression genetic prediction models were established using complementary methods of ENET (Zou and Hastie, 2005), LASSO (Baselmans et al., 2019), and MCP (Zhang, 2010), which have been widely used in TWASs to predict genetically regulated expression levels (Schaid et al., 2018). For each model, we considered single nucleotide polymorphisms (SNPs) within the gene's cis-regulatory (within a 1-Mb window of the gene's transcription site) or enhancer–promoter interaction regions as candidate predictors. The enhancer–promoter interaction regions were determined by GenHancer (Fishilevich et al., 2017), an existing comprehensive database that integrates reported enhancers from four genome-wide resources, including ENCODE, Ensembl, FANTOM, and VISTA. To determine the best-performing prediction model, we compared the R2 values of the models yielding a satisfactory prediction accuracy, that is, R2 > 0.01. The models for 13,427 splicing introns in total were used in further analyses for PCa-risk associations.
Association analyses of predicted splicing intron expression with PCa risk
Associations of genetically predicted splicing intron expression with PCa risk were investigated using the summary statistics generated from 79,194 PCa cases and 61,112 controls of European ancestry included in the PRACTICAL, CRUK, CAPS, BPC3, and PEGASUS consortia (Schumacher et al., 2018; Wu et al., 2019a). The detailed information for this data set has been described elsewhere (Wu et al., 2020; Wu et al., 2019b).
In brief, 46,939 cases and 27,910 controls were genotyped using OncoArray with 570,000 SNPs (http://epi.grants.cancer.gov/oncoarray). The data from several previous PCa GWASs of European ancestry: UK stage 1 (1854 cases and 1894 controls) and stage 2 (3650 cases and 3940 controls), CaPS 1 (478 cases and 428 controls) and CaPS 2 (1458 cases and 512 controls), BPC3 (2068 cases and 2993 controls), NCI PEGASUS (4600 cases and 2941 controls), and iCOGS (20,219 cases and 20,440 controls), were also included. Using an inverse variance fixed-effect method, logistic regression summary statistics was then meta-analyzed.
The associations of predicted splicing intron expression with PCa risk were further estimated using intron expression prediction weights, summary statistics of SNP-PCa risk associations, and an SNP-correlation (linkage disequilibrium [LD]) matrix with 1000G EUR population as reference, using the S-PrediXcan framework (Gusev et al., 2016).
The false discovery rate (FDR)-corrected p-value threshold was used to determine significant associations between genetically predicted splicing intron expression and PCa risk.
Fine-mapping analyses to prioritize putatively causal splicing introns
Fine-mapping of causal gene sets (FOCUS) fine-mapping analyses of the identified associated splicing introns were performed to prioritize the most likely causal splicing introns for PCa risk, as described elsewhere (Mancuso et al., 2019). In summary, three files were used to run FOCUS fine-mapping analysis, including sQTL weights from each corresponding splicing intron prediction model, GWAS summary statistics for PCa risk, and LD estimated from plink-formatted 1000G.EUR.QC. The default 90%-credible splicing intron set was used to identify the most likely causal splicing introns after FOCUS outputted the posterior probability for each splicing intron.
Functional enrichment analysis
“Canonical Pathway,” “Disease and Functions,” and “Network” function of the “Core Analysis” in Ingenuity Pathway Analysis (IPA) (Krämer et al., 2014) were performed to assess the enriched pathways, functional categories, and networks of the identified PCa risk genes. For significantly enriched “Canonical Pathway,” the significant p-value was calculated using the right-tailed Fisher's exact test. “Disease and Functions” displayed the annotated biological function and/or linked diseases of the genes. The “Network” function was constructed based on the Ingenuity Knowledge Base from published studies.
Ethics approval
This study used deidentified data and no ethics approval or consent was required.
Data Availability
The PCa GWAS summary statistics in the PRACTICAL consortium is available at http://practical.icr.ac.uk/blog/?page_id=8164 The full association results from our analyses and codes are available upon request.
Results
Splicing intron expression genetic prediction models
In this study, three sets of splicing intron expression prediction models developed using three complementary methods, namely, ENET, LASSO, and MCP models, were used to detect the associations of predicted expression of splicing introns with PCa risk. There are satisfactory prediction models for 4366 splicing introns using the ENET method, 3812 for LASSO, and 5249 for MCP, which were tested for their associations with PCa risk.
Associations of predicted expression of splicing introns with PCa risk
For each splicing intron with two or three expression prediction models developed, only the model showing the highest prediction performance was used to assess the splicing intron of interest. Based on the association analyses, 120 splicing introns of 97 genes were significantly associated with PCa risk at FDR-corrected threshold p < 0.05. Of them, 38 splicing introns of 31 genes were associated at the Bonferroni-corrected threshold p < 4.90 × 10−7 (0.05/10,207), after excluding 49 splicing introns of 35 genes located in LD-extensive regions (Fig. 1 and Table 1 and Supplementary Tables S1–S3). Of the 120 splicing introns, the higher predicted expression of 63 splicing introns was associated with increased PCa risk. Inversely, the lower predicted expression of 57 splicing introns was associated with increased PCa risk.

Manhattan plot of association results from the prostate cancer transcriptome-wide association study using splicing intron expression genetic prediction models. The x axis represents the genomic position of each tested splicing intron, and the y axis represents Z value of the association. Each dot represents the genetically predicted expression of one specific splicing intron. The upper dark line represents p = 9.96 × 10−7 for the Bonferroni correction threshold (0.05/50,220), and the lower gray line represents p = 7.53 × 10−4 for the false discovery rate-corrected threshold.
Associations of 35 Splicing Introns of 34 Genes That Have Not Been Reported in Previous Transcriptome-Wide Association Study or Splicing Transcriptome-Wide Association Study of Prostate Cancer Risk and at Novel Loci That Have Not Been Reported in Genome-Wide Association Studies of Prostate Cancer Risk
Protein, protein coding genes; lncRNA; other, transcribed unprocessed pseudogene.
Number of SNPs among “SNPs” located in either gene-body region or enhancer–promoter region and used to construct nonzero weights.
Bonferroni-corrected significant introns shown in bold.
The closest risk SNPs identified in previous GWASs for PCa risk.
ENET, elastic net; FDR, false discovery rate; GWAS, genome-wide association study; LASSO, least absolute shrinkage and selection operator; lncRNA, long noncoding RNA; MCP, minimax concave penalty; PCa, prostate cancer; SNPs, single nucleotide polymorphisms.
Of the identified 120 splicing introns, 67 locate in 46 genes that have been reported in previous TWASs (Supplementary Table S1) and/or PCa spTWASs (Supplementary Table S2). Also, 18 splicing introns of 17 genes were at genomic loci within 500 kb of GWAS-identified PCa risk variants while have not yet been reported in previous TWASs or spTWASs (Supplementary Table S3).
Importantly, 35 splicing introns of 34 novel genes were identified to be associated with PCa risk for the first time (Table 1). These 34 genes include 32 protein-coding genes (TCEA3, XKR8, SNAP47, THAP4, HEMK1, CNBP, PRKAR1B, GTPBP10, LRGUK, HABP4, LCN2, PNPLA2, SCYL1, LOXL1, DNAJA3, ANKS3, RAI1, SHMT1, ACACA, DDX42, ZADH2, SSBP4, U2AF1L4, ARHGAP33, PLD3, B9D2, BCKDHA, DMPK, NTN5, SNTA1, PCBP3, and SLC2A11), 1 transcribed unprocessed pseudogene (NSUN5P2), and 1 long noncoding RNA (lncRNA; MIR22HG). Of these, 16 genes have already been reported to be related to PCa risk in previous studies, including TCEA3, SNAP47, THAP4, HEMK1, PRKAR1B, GTPBP10, LCN2, PNPLA2, RAI1, ACACA, U2AF1L4, ARHGAP33, B9D2, BCKDHA, DMPK, and PCBP3 (Supplementary Table S4) (Boutros et al., 2015; Brikun et al., 2018; Chen and Hu, 2019; Cheung et al., 2017; Ding et al., 2015; Jin et al., 2016; Jo et al., 2017; Martinez-Marin et al., 2017; Roussigne et al., 2003; Schroder et al., 2022; Shahabi et al., 2016; Svensson et al., 2014; Wang et al., 2020; Zahalka et al., 2017; Zhang et al., 2022; Zhao et al., 2020).
Of the identified novel splicing introns, an association between higher predicted expression and increased PCa risk was observed for 22 splicing introns. Conversely, an association between lower predicted expression and increased PCa risk was detected for 13 introns.
Fine-mapping analysis results
Of the identified 120 splicing introns of 97 genes associated with PCa risk, 21 splicing introns of 19 genes were further prioritized by FOCUS analysis (Table 2), with 90%-credible sets to be likely causal splicing introns (Mancuso et al., 2019). Of them, two splicing introns (chr16:4446084-4446886:DNAJA3 and chr16:4698941-4699052:ANKS3) of two genes (DNAJA3 and ANKS3) were reported for the first time.
Twenty-One Splicing Introns of 19 Genes Prioritized by Fine-Mapping of Causal Gene Sets Fine Mapping Analysis to Be Putatively Causal Units
TWAS, transcriptome-wide association study.
IPA results
“Core Analysis” function of IPA (version 01-20-04; Ingenuity System, Inc.), including “Canonical Pathway,” “Disease and Functions,” and “Network” analyses, was performed for the 97 genes identified in our spTWAS. For the “Disease and Functions,” we mainly focused on cancer, lipid metabolism, and PCa-related diseases and function categories. Twenty-two genes (ABHD12, ACACA, ADAM15, BCKDHA, CASP8, CREB3L4, FAAH, FARP2, GNB1L, HIBADH, MKNK1, NTN5, PLD3, PNPLA2, PRKAR1B, PTGR3, SEC61A1, SEC61A2, SHMT1, SIRT2, VAMP8, and YWHAQ) enriched in 32 canonical pathways are shown in Supplementary Table S5. These canonical pathways contain triacylglycerol degradation (enriched genes ABHD12, FAAH, and PNPLA2; p = 1.66 × 10−3) and autophagy (enriched genes CREB3L4, GNB1L, PRKAR1B, and VAMP8; p = 1.07 × 10−2). Previous research has supported that autophagy can regulate lipolysis and cell survival by degrading lipid droplets in PCa cells (Kaini et al., 2012).
Based on the “Disease and Functions” analysis, 89 identified genes were enriched for cancer, lipid metabolism, and 2 PCa-related functional categories (p < 0.05) (Supplementary Table S6). Functions of 89 associated genes were related to cancer, 16 were related to lipid metabolism, and 33 were related to PCa. Except for NSUN5P2, MIR22HG, and ZADH2, 31 novel genes (TCEA3, XKR8, SNAP47, THAP4, HEMK1, CNBP, PRKAR1B, GTPBP10, LRGUK, HABP4, LCN2, PNPLA2, SCYL1, LOXL1, DNAJA3, ANKS3, RAI1, SHMT1, ACACA, DDX42, SSBP4, U2AF1L4, ARHGAP33, PLD3, B9D2, BCKDHA, DMPK, NTN5, SNTA1, PCBP3, and SLC2A11) were enriched in cancer categories (p = 3.29 × 10−2–9.97 × 10−6). Reassuringly, nine of them, namely, SNAP47, LCN2, SCYL1, DNAJA3, RAI1, DDX42, PLD3, BCKDHA, and DMPK, were also enriched in the PCa functional category (p = 1.17 × 10−2).
Based on the “Network” analysis, six networks of spTWAS-identified genes (Score ≥13) and three networks of spTWAS-identified novel genes were identified (Score ≥15) (Supplementary Table S7 and Supplementary Figs. S1 and S2). Interestingly, 11 novel PCa susceptibility genes (ACACA, BCKDHA, DNAJA3, GTPBP10, HABP4, LCN2, LOXL1, NSUN5P2, PLD3, PNPLA2, and SLC2A11) were enriched in a cancer-related network: cancer, dermatological diseases and conditions, organismal injury, and abnormalities (score = 25, Fig. 2). Five genes, namely, ACACA, DNAJA3, LCN2, LOXL1, and PNPLA2 were in the node of this network.
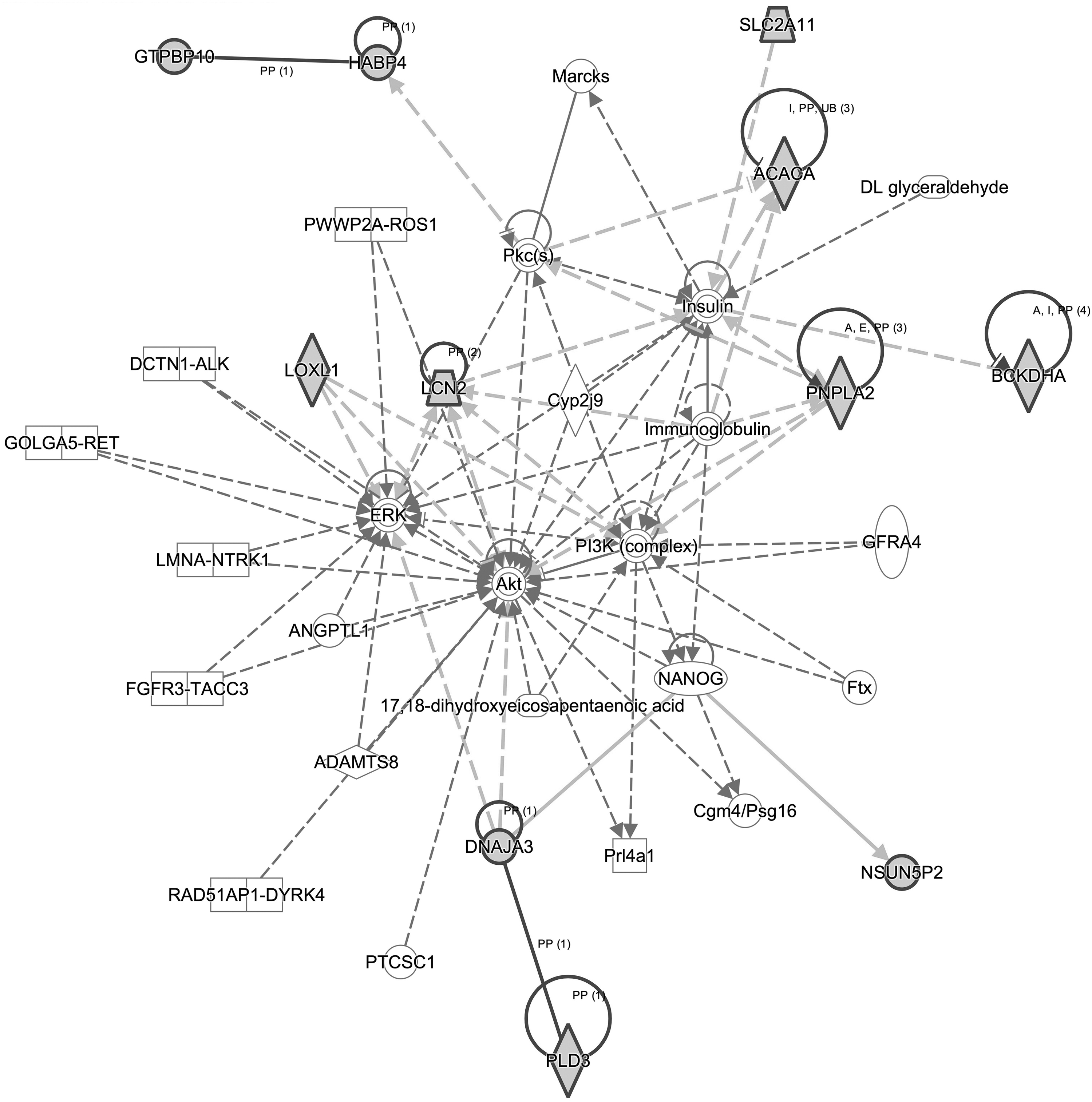
Splicing TWAS identified 11 novel genes enriched in a cancer-related network. Eleven novel genes were highlighted in the solid dark line. TWAS, transcriptome-wide association study.
Discussion
In this study, we conducted a comprehensive spTWAS to identify candidate splicing introns with genetically predicted expression to be associated with PCa risk. The spTWAS analysis identified 97 genes, including 34 novel genes that were not reported in any previous TWAS and/or spTWAS (Emami et al., 2019; He et al., 2022; Liu et al., 2022; Mancuso et al., 2018; Wu et al., 2019b). The identified genes were enriched in triacylglycerol degradation and autophagy pathways, and lipid metabolism, cancer, and PCa functional categories, which provides novel knowledge for better understanding the etiology of PCa.
Some of the novel genes identified in our study have already been reported in the literature as potentially playing a role in PCa development. For example, TCEA3 encodes transcription elongation factor A3, which is a downregulated protein in PCa tissue, and the minor intron splicing efficiency of TCEA3 could increase the development of lethal PCa (Augspach et al., 2021). Another gene ACACA encodes acetyl-CoA carboxylase alpha, which is one of the rate-limiting enzymes for fatty acid synthesis (Shafi et al., 2015). Compared with normal tissue, high expression of ACACA in PCa tumor tissue was reported in PCa (Swinnen et al., 2000). As a result, downregulation of ACACA could inhibit the malignant progression of PCa (Zhang et al., 2021). Further functional studies are needed to better characterize the potential roles of these genes and splicing introns in prostate tumorigenesis.
In PCa cells, lipid degradation can promote cell survival (Itkonen et al., 2017) by providing a main bioenergetic pathway through fatty acid oxidation (Liu, 2006). In “Canonical Pathway” analysis, the top enriched canonical pathway was triacylglycerol degradation. Three genes (ABHD12, FAAH, and PNPLA2) were enriched in this canonical pathway. Besides these 3 genes, other 13 genes (ACACA, ACTN4, CASP8, CERS2, CNBP, LCN2, MKNK1, NPNT, PIBF1, SIRT2, SLC45A3, THADA, and XKR8) involved in lipid metabolism were uncovered by “Disease and Functions” analysis. These genes were involved in triacylglycerol degradation (such as ABHD12, FAAH, and PNPLA2), fatty acid synthesis (such as ACACA), and lipid transport (such as LCN2). The above results suggested that lipid metabolism might play a key role in PCa progression (Wu et al., 2014).
Of the identified genes, most (89/97) were implicated in cancer. It should be emphasized that the functions of nine novel genes (SNAP47, LCN2, SCYL1, DNAJA3, RAI1, DDX42, PLD3, BCKDHA, and DMPK) had been linked to PCa. Moreover, 11 novel PCa susceptibility genes were enriched in a cancer-related network. In particular, five genes (ACACA, DNAJA3, LCN2, LOXL1, and PNPLA2) were in the node of this network, implicating potential roles of these genes in PCa development. The splicing introns of the abovementioned genes could potentially serve as candidate markers for PCa risk, and further studies are needed to better characterize them.
Three methods for developing splicing expression genetic prediction models, namely, LASSO, MCP, and ENET, were leveraged in the current study. This provides a valuable opportunity to detect PCa-associated splicing introns with different genetic regulatory mechanisms. LASSO, MCP, and ENET are popular penalization methods, each of which performs optimally under different genetic architectures. To further enhance the prediction accuracy, we incorporated enhancer regions as they play key regulatory roles in splicing (Lee and Rio, 2015). Our study leveraging these complementary models can lead to more powerful discovery.
There are several strengths of the current study. We leveraged splicing expression prediction models of normal prostate tissue instead of prostate tumor tissue. For many introns, the splicing expression patterns change during tumor development (Ghigna et al., 2008). The directions of associations for a large proportion of the identified associated splicing introns were consistent across the ENET, LASSO, and MCP models (Supplementary Table S8), indicating that our findings are robust. The strategies we used helped to identify more genes than those identified in He et al. (2022), in which a splicing susceptible transcription factor (sTF) TWAS was conducted and six novel genes (NOL10, WTAP, BRI3, USP39, SNRPC, and CCND1) were reported.
On the contrary, several potential limitations need be considered for appropriately the interpretating results of the present study. One limitation is that the identified associations may not necessarily imply causality. This is a limitation known to exist for the conventional TWAS design (Wainberg et al., 2019). Future functional investigation would be needed to better understand whether the identified splicing introns and genes play a causal role in prostate tumorigenesis. Another limitation is that we were not able to evaluate whether the associations of our identified splicing introns differ according to family history of PCa and tumor stage/grade due to a lack of relevant information. Additional work investigating these is needed to better understand their relationship. A third limitation is that other potential factors, such as smoking, alcohol consumption, obesity, and family history of PCa, were not included in our model building, which may confound our association results.
Further study is needed to better evaluate the potential influence of such factors in detecting PCa-related splicing introns.
Conclusions
In summary, we performed a comprehensive spTWAS and identified novel susceptibility splicing introns for PCa risk. We detected associated splicing introns located in 34 novel genes for PCa risk. Our study identifies novel genes and splicing introns for PCa risk and our findings improve the etiology understanding of this common malignancy.
Footnotes
Authors' Contributions
L.W. and C.W. conceived and jointly supervised the study. Y.S., Y.E.B., and J.Z. contributed to the study design, performed statistical analyses, and wrote the article. Z.Z., H.Z., and C.C. contributed to data analysis and result interpretation. All authors contributed to the article revision and approved the final article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study is supported by the University of Hawaii Cancer Center, and the Teacher Training Project of Longyan University, and the Provincial Key Science and Technology Project jointly funded by the Fujian Provincial Department of Industry and Information and the Fujian Provincial Department of Education, China (grant 2021G02015).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.