
Abstract
Idiopathic pulmonary arterial hypertension (IPAH) is a progressive disease that affects the pulmonary arteries, resulting in increased pulmonary vascular resistance and right ventricular dysfunction, which can ultimately lead to heart failure and death. The molecular substrates of IPAH are poorly understood while diagnostics and therapeutics innovation remain as unmet needs for this debilitating disease. In this study, a network-based methodology was used to uncover the salient molecular mechanisms of IPAH to inform drug and diagnostic discovery, and personalized medicine. Expression profiling datasets associated with IPAH were obtained from the Gene Expression Omnibus database: GSE15197, GSE113439, GSE53408, and GSE67597. The comparative analysis of mRNA and miRNA expression data and the modular analysis of a transcriptome-based weighted gene coexpression network unraveled disease-specific gene and miRNA signatures. DEAD-box helicase 52 (DDx52), ESF1 nucleolar pre-RNA processing protein (ESF1), heterogeneous nuclear ribonuclearprotein A3 (MNRNPA3), Myosin VA (MYO5A), replication factor C subunit 1 (RFC1), and arginine and serine rich coiled coil 1 (RSRC1) were detected as the salient genes for IPAH. In addition, the salient gene-based drug repositioning analysis identified alvespimycin, tanespimycin, geldanamycin, LY294002, cephaeline, digoxigenin, lanatoside C, helveticoside, trichostatin A, phenoxybenzamine, genistein, pioglitazone, and rosiglitazone as potential drug candidates for IPAH. In conclusion, this study provides new molecular signatures in relation to IPAH and attendant potential drug candidates for further experimental and translational clinical research for patients with IPAH.
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a chronic and progressive disease characterized by elevated pulmonary artery pressure leading to right heart failure and death (Schlueter et al., 2020; Swaminathan et al., 2020). An increase in the mean pulmonary artery pressure >25 mmHg at rest, a pulmonary vascular resistance of greater than three Wood units, and a pulmonary capillary wedge pressure of <15 mmHg at end expiration are used to identify the disease (Luo et al., 2020). IPAH is the sporadic form of pulmonary arterial hypertension while familial form of the disease caused by inherited genetic mutations also exists as well as secondary forms owing to drugs and connective tissue disease (Rajkumar et al., 2010; Zhao et al., 2014).
The prevalence of pulmonary arterial hypertension (PAH) and IPAH is 15 and 5.9 cases per million adult population, respectively, according to the 2015 European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines (Galiè et al., 2015). Hendriks et al. reported a 5- and 10-year transplant-free survival rates of 71% and 35%, respectively. Despite advances in treatment strategies, the survival rate for incident patients still remains unsatisfactory (Hendriks et al., 2022).
The pathophysiological changes associated with PAH are diverse and complex. Several genetic, epigenetic, and environmental factors contribute to its progression. Proliferative remodeling, endothelial cell dysfunction, fibrosis, and inflammation are among the main pathological hallmarks of PAH (Li et al., 2020; Mayeux et al., 2021). PAH also exhibits cancer-like biological features such as cell proliferation, angiogenesis, and resistance to apoptosis of fibroblasts, pulmonary artery smooth muscle cells, and pulmonary artery endothelial cells (Boucherat et al., 2017; Wu et al., 2016; Zeng et al., 2021).
Although the impact of PAH has been widely reported, its pathogenesis is still not fully understood (Li et al., 2020). Therefore, the elucidation of the molecular mechanisms underlying PAH would improve our understanding of the disease pathogenesis and provide valuable information for the discovery and development of predictive biomarkers.
Therapeutic options for patients with PAH have gradually evolved over the past decade. Treatment strategies aim to improve patients' quality of life, exercise capacity and right ventricular function, and reduce the risk of mortality (Galiè et al., 2015). Medical management includes drugs that target pathways associated with disease progression, such as the nitric oxide, endothelin-1, and prostacyclin pathways (Mayeux et al., 2021). The beneficial effects of anticancer drugs in combination with vasodilators have also been investigated (Boucherat et al., 2017). Current therapies can alleviate PAH by reducing the pulmonary vascular resistance, but no effective drug therapy has been developed to completely reverse the vascular remodeling of the pulmonary arteries (Galiè et al., 2015; Li et al., 2020). Therefore, new approaches for the discovery of molecular targets and drug candidates are needed.
The development of omics technologies enables system-level network analysis to unravel the molecular mechanisms behind human diseases and to identify novel biomarker candidates for therapeutic purposes. A multi-omics approach to PAH may lead to the identification of new diagnostic and therapeutic targets and improve treatment efficacy.
Weighted gene coexpression network analysis (WGCNA) identifies highly correlated gene modules, determines modules significantly associated with sample traits, and identifies hub genes using module-to-module and gene-to-module relationship measures (Langfelder and Horvath, 2008). Application of WGCNA to various human diseases has led to the identification of important molecular targets for treatment and therapeutic purposes (Auwul et al., 2021; Brohawn et al., 2016; Yan, 2018; Zhang et al., 2018a).
In this study, an integrative analysis of genome-wide gene and miRNA expression data from patients with IPAH and controls was performed to identify molecular signatures and repositioned drug candidates for therapeutic purposes. Specifically, differentially expressed genes (DEGs), miRNAs (DEmiRNAs), and their associations were identified. In addition, a gene coexpression network was constructed, and co-analyzed with human protein-protein interaction (PPI) network to identify novel molecular signatures. Following the cross-validation analysis, a drug repositioning approach was applied to determine the potential drugs that could be effective for IPAH.
Materials and Methods
Study sample, data acquisition, and analysis
Expression profiling datasets associated with IPAH were obtained from the Gene Expression Omnibus (GEO) database (Barrett and Edgar, 2006) at the National Center for Biotechnology Information.
The explore dataset GSE15197 (Rajkumar et al., 2010), which was performed based on the Agilent-014850 Whole Human Genome Microarray 4x44K G4112F platform, contained 39 lung samples from 18 patients with IPAH, 8 patients with idiopathic pulmonary fibrosis (IPF) with secondary IPAH, and 13 normal controls. As IPAH was specifically investigated in this study, the samples from IPF with secondary IPAH were excluded from that dataset.
The datasets of GSE113439 (Mura et al., 2019) and GSE53408 (Zhao et al., 2014) were downloaded for further data validation analysis. GSE113439 contains lung samples from 6 patients with IPAH and 11 normal controls; and GSE53408 contains lung samples from 5 patients with IPAH and 11 normal controls based on the Affymetrix Human Gene 1.0 ST Array platform. PAH secondary to other diseases were excluded from the validation datasets and only IPAH samples were used for further analyses. The miRNA expression profiling dataset GSE67597 (Wu et al., 2016), obtained from the Agilent-046064 Unrestricted_Human_miRNA_V19.0_Microarray platform, consisted of lung tissues samples from 15 individuals, including 7 IPAH patients and 8 age-matched control donors.
Differential gene expression and miRNA expression analyses were performed using the GEO2R tool (www.ncbi.nlm.nih.gov/geo/geo2r/). Raw gene and miRNA expression data were normalized by quantile normalization, and DEGs and DEmiRNAs were identified using normalized expression values by Linear Models of Microarray Data (LIMMA) (Smyth, 2004). The Benjamini–Hochberg method was used to control the false discovery rate (FDR). Statistical significance and expression patterns were determined by adjusted p-value (adj. p-value) and fold-change (FC), respectively. An adjusted p-value threshold of 0.05 (adj. p-value <0.05) was maintained to identify significantly expressed genes (SEGs). Then, an FC cutoff of 2 was used to determine the regulatory pattern (up- and downregulation) of each gene to identify DEGs. A p-value threshold of 0.05 (p < 0.05) and an FC cutoff of 4 were used to determine the statistical significance and regulatory pattern of each miRNA, respectively.
To elucidate the association between DEmiRNAs and DEGs, experimentally validated miRNA-target gene interactions were retrieved from the miRTarbase database (Release 9.0) (Huang et al., 2022).
This article presents a computational analysis of gene expression data obtained from public databases. It does not contain any studies with human and animal subjects and therefore does not require IRB approval.
Construction of weighted gene coexpression network and selection of key modules
The weighted gene coexpression network was constructed using the WGCNA package (Langfelder and Horvath, 2008) version 1.70–3 in R software (R Core Team, 2020). The samples in the GSE15197 dataset were clustered based on the average linkage hierarchical clustering method through the hclust function, and the cluster dendrogram was constructed to check and exclude the outlier samples.
For network construction, soft-threshold power β was determined by the pickSoftThreshold function according to the standard of scale-free networks, and correlations between all gene pairs were calculated by Pearson correlation coefficients (PCCs). First, a signed adjacency matrix was constructed using the soft-threshold power and the similarity matrix consisting of the PCCs of all gene pairs. Then, the adjacency matrix was transformed into the topological overlap matrix (TOM), and the dissimilarity matrix (dissTOM) was computed.
The coexpression modules were then determined using the dynamic branch-cutting method, with the minimum module size and deepSplit set to 30 and 2, respectively. Genes with similar expression profiles were placed in the same modules and a hierarchical clustering dendrogram of genes was constructed. Similar modules were further merged by using module eigengene (ME), which summarizes the expression profiles of each module. The dissimilarity of the MEs was calculated and the ME cut height was set to 0.25 as the module merging criterion.
Key modules that are significantly associated with clinical traits of IPAH were determined by calculating the PCCs between MEs and clinical traits. Specifically, modules that showed significant positive correlation with IPAH (r > 0.6, and p-value <0.01) were selected as key modules. Then, genes in key modules were prioritized based on their connectivity, and the highly connected genes were determined using two measures: gene significance (GS) and module membership (MM). GS, which shows the correlation between gene expression and clinical trait, and MM, which defines the correlation between gene expression and ME, were calculated for each gene in key modules.
Functional enrichment analyses
Functional enrichment analysis was performed using the ShinyGO (v0.76.3) enrichment tool (Kamburov et al., 2013) to identify significantly enriched Gene Ontology (GO) biological process (BP) terms and molecular pathways using the GO (Ashburner et al., 2000) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) databases as annotation sources, respectively. Enrichment analysis was calculated based on the hypergeometric distribution and p-values were corrected using FDR. An FDR cutoff of 0.05 (FDR <0.05) was used to determine the statistical significance.
Reconstruction and analysis of PPI networks
PPIs between the proteins encoded by coexpressed genes were extracted from the STRING database (https://string-db.org) (Szklarczyk et al., 2021) using a confidence score of 0.7. PPI networks were reconstructed individually for each key module via Cytoscape version 3.9.1 (Shannon et al., 2003). Network modules were identified by Cytoscape's MCODE plug-in (Bader and Hogue, 2003). In MCODE, network scores were computed by excluding the loops. Modules were identified by setting the degree threshold, node score threshold, K-core threshold, and maximum depth to 2, 0.2, 2, and 100, respectively. The fluff parameter was turned off and the haircut parameter was turned on.
Determination and validation of hub genes
To increase the biological significance and to better represent the genetic characteristics of IPAH, hub genes were identified by a combined analysis of WGCNA key modules, PPI network modules, and DEGs. In a coexpressed gene module, hub genes were determined taking into account their correlation with the clinical trait and the degree of connectivity. Therefore, GS and MM values were calculated, and genes that fulfilled |MM| > 0.8 and |GS| > 0.5 in key modules were selected as WGCNA hub genes. In PPI networks, hub proteins were determined based on modularity. The genes encoding proteins in modules with at least 5 members and a minimum score of 5 were recognized as PPI hub genes. The common genes among the WGCNA hubs, PPI hubs, and DEGs of the GSE15197 dataset were selected for validation analyses.
Two independent gene expression datasets, GSE113439 and GSE53408, were used to test the validity of the common hub genes. The genes that also showed significantly altered expression in the GSE113439 and GSE53408 datasets were selected as key genes for further analysis.
Diagnostic performance analysis of key genes
Key genes were subjected to diagnostic performance analysis. Gene expression profiles of the key genes were extracted from the GSE15197 dataset, and receiver operating characteristic (ROC) curves were plotted in R. The area under the curve (AUC), which represents sensitivity (the proportion of positive test results in patients) and specificity (the proportion of negative test results in healthy individuals), was used to evaluate the diagnostic performance of the key genes. Statistical significance was determined using an AUC value threshold of 70% and genes with an AUC value ≥70% were considered as diagnostic.
Identification of candidate drugs
Drug Gene Budger (DGB) (Wang et al., 2019b), a web-based application, was searched for potential therapeutic agents targeting key genes identified in IPAH. DGB provides a list of small molecules that are predicted to affect the expression of the target gene of interest. The expression patterns (upregulation or downregulation) of target genes in response to the predicted small molecules were assessed using the log-transformed FC, p-value, and q-value for each small molecule, and the significance of differential expression was determined using the LIMMA method.
The experimental data extracted from the original Connectivity Map (Lamb et al., 2006) and GEO datasets compiled by the DGB were used to identify drugs or compounds targeting key genes. Significant drugs were selected based on a q-value threshold of 0.05 (q-value <0.05). The repositioned drugs were further investigated through L1000 fireworks display (L100FWD) (Wang et al., 2018) and Drugbank (Wishart et al., 2018) databases, and unknown drugs were eliminated.
Results
DEGs and DEmiRNAs in IPAH
The explore dataset (GSE15197) containing gene expression data from lung samples of IPAH patients and healthy controls was comparatively analyzed to identify DEGs and their expression patterns. Statistical analyses revealed 7466 SEGs based on an adj. p-value <0.05 criterion. Considering the FC in expression levels, a total of 2361 DEGs (1254 upregulated, 1107 downregulated) were identified (Fig. 1A).

Global miRNA expression profile analysis was performed using the GSE67597 dataset. A total of 51 miRNAs showed significantly altered expression levels in IPAH compared with controls. In addition to p-values, at least an 4-FC in mean expression levels was used to identify DEmiRNAs and their regulatory pattern. This analysis revealed 12 upregulated (miR-505-5p, miR-183-5p, miR-375, miR-500a-3p, miR-6074, miR-152, miR-31-5p, miR-198, miR-648, miR-4473, miR-3974, and miR-205-5p) and 9 downregulated (miR-146b-3p, miR-1256, miR-302f, miR-517a-3p, miR-4693-5p, miR-4704-3p, miR-1178-3p, miR-495-5p, and miR-1253) DEmiRNAs (Fig. 1B).
To reveal regulatory associations in disease pathogenesis, the miRNA-target gene regulatory network was reconstructed by collecting DEmiRNA-DEG interactions. The constructed network contained 423 associations between 343 DEGs and 20 DEmiRNAs (Supplementary Fig. S1). To elucidate the molecular pathways and BPs underlying IPAH, functional enrichment analysis of DEmiRNAs was performed via DEGs regulated by these DEmiRNAs (Supplementary Fig. S2).
Pathway enrichment of upregulated DEmiRNAs revealed several cancer and cancer-associated pathways, several infection-related disease pathways, MAPK signaling, HIF-1 signaling, neurotrophin signaling, sphingolipid signaling, lipid and atherosclerosis, hypertrophic and arrhythmogenic right ventricular cardiomyopathy, ferroptosis, and endocrine-related pathways such as endocrine resistance and prolactin signaling. In addition, RNA processing, neurogenesis, neuron generation, central nervous system development, glial cell development, transmembrane transport,
Weighted gene coexpression network
Gene coexpression network analysis was performed on 7466 SEGs from the explore dataset GSE15197. The cluster dendrogram of the samples revealed one sample (GSM379341) as an outlier (Fig. 2A). Therefore, this sample was excluded from further analysis, and 30 samples (17 IPAHs, and 13 controls) were included for WGCNA. A scale-free network was constructed with a soft-threshold power of β = 20 and R2 = 0.90 (Fig. 2B). A total of 21 modules were identified (Fig. 2C). The genes with similar expression profiles were grouped into the same module, and after module merging, three coexpressed modules were obtained. The cluster dendrogram of these modules is given in Figure 2C. The magenta module containing 2753 genes was the largest module, followed by the black module (1623 genes) and the brown module (1589 genes).
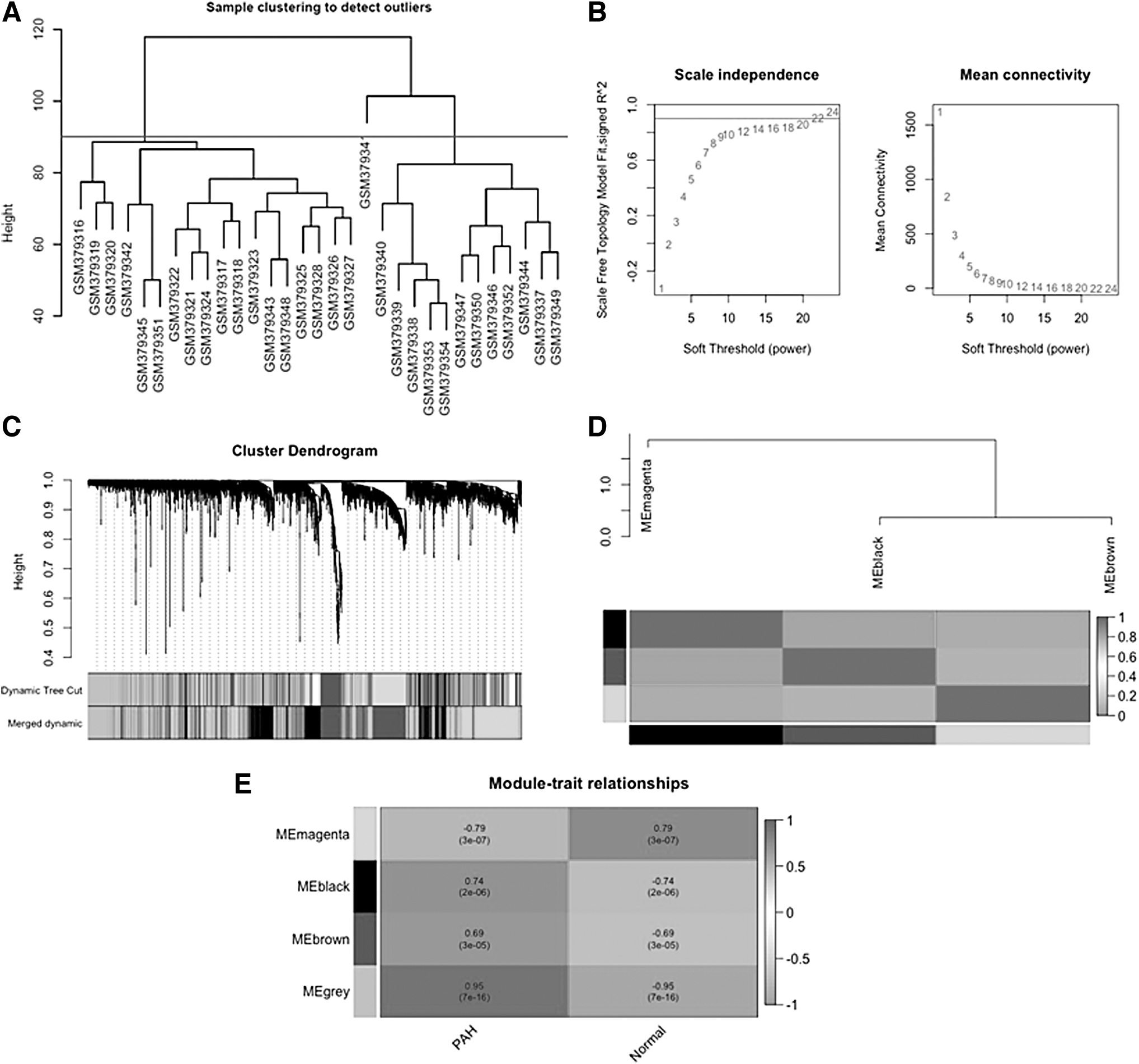
Construction of the WGCNA network.
All ungrouped genes that were not coexpressed were included in the gray module (1501 genes). The correlations between the coexpressed modules were presented using the ME dendrogram and heatmap (Fig. 2D).
To better understand the biological significance of coexpressed gene modules in disease progression, the correlation between MEs of major modules and clinical traits was considered (Fig. 2E). The module–trait relationship analysis showed that the black module (r = 0.74, p-value = 2E-06) and the brown module (r = 0.69, p-value = 3E-05) had a significant positive correlation with IPAH. Since genes in the black and brown modules may play a key role in the progression of IPAH, these modules were selected as the clinically significant key modules.
To gain further biological insight into the genes in the brown and black modules, enrichment analyses were performed, and the top 10 significantly enriched GO BP terms, and KEGG pathways are given in Figure 3.
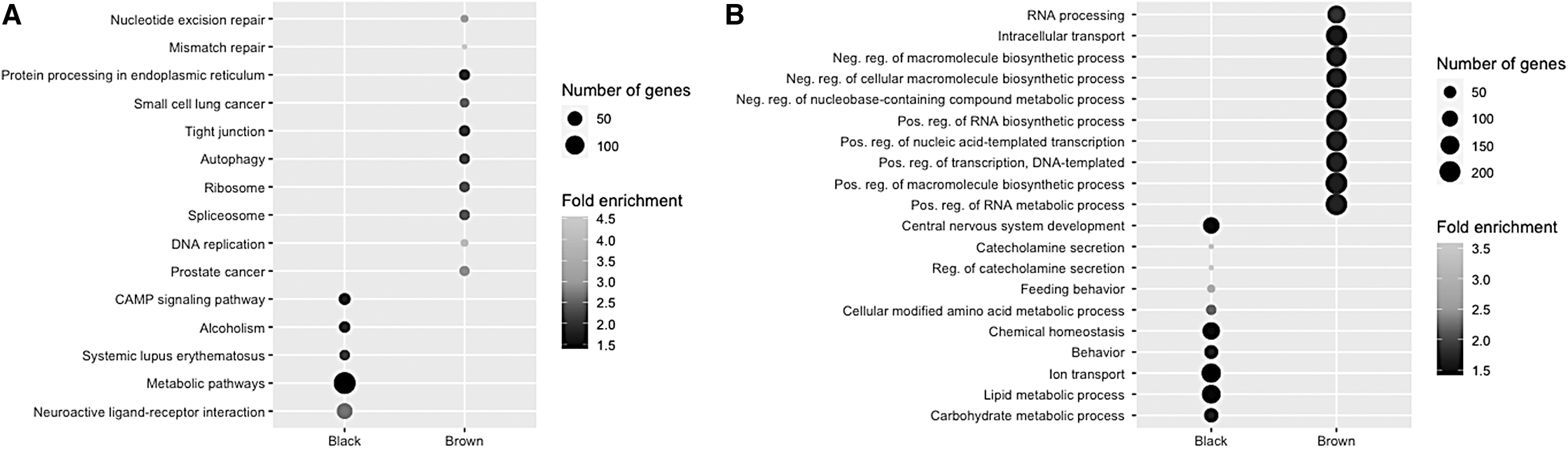
Functional enrichment analysis of key module genes.
Pathway enrichment analysis revealed that the brown module genes were mainly enriched in pathways involved in maintaining genome stability (mismatch repair, nucleotide excision repair, and spliceosome), pathways involved in protein processing (ribosome, and protein processing in endoplasmic reticulum), several cancer pathways, various infectious diseases, autophagy, HIF-1 signaling pathway, and EGFR tyrosine kinase inhibitor resistance. In addition, brown module genes were found to be significantly involved in RNA processing, chromatin organization and remodeling, histone modification, methylation, gene silencing, cell cycle, neurogenesis, brain development, and neuron recognition.
The genes in the black module were significantly enriched for neuroactive ligand–receptor interaction, systemic lupus erythematosus, and CAMP signaling pathway. Carbohydrate and lipid metabolism, catecholamine secretion, nervous system development, forebrain development, and synaptic signaling were found among the significantly enriched BPs in this module.
Identification and validation of hub genes
The hub genes of key modules were explored based on GS and MM values to better represent the clinical signatures, and a total of 1550 genes (|MM| > 0.8 and |GS| > 0.5) were identified as WGCNA hubs. Furthermore, PPI networks were reconstructed around the genes in key modules, and the modular analyses of the black PPI network consisted of 1367 interactions between 643 proteins, and the brown PPI network consisted of 3146 interactions between 895 proteins, revealed a total of 205 PPI hubs (63 and 142 hubs in the black and brown PPI networks, respectively). The intersection of WGCNA hubs, PPI hubs, and DEGs of the GSE15197 dataset were then selected as core genes. The 38 commonly identified core genes were further analyzed.
Two independent gene expression datasets, GSE113439 and GSE53408, were used to validate the core genes as candidate biomarkers for IPAH. Among these genes, DDX52, ESF1, HNRNPA3, MYO5A, RFC1, and RSRC1 were also found to be differentially expressed in both GSE113439 and GSE53408 datasets (Supplementary Table S1), and were therefore considered as key gene signatures in IPAH.
Key genes included DEAD-box helicase 52 (DDX52), nucleolar pre-rRNA processing protein (ESF1), and heterogeneous nuclear ribonucleoprotein A3 (HNRNPA3), which enable RNA binding activity. Myosin VA (MYO5A), which is a member of the myosin gene superfamily, has functions in melanosome transport, and mediates the transport of vesicles to the plasma membrane. Replication factor C subunit 1 (RFC1) encodes the large subunit of replication factor C, and acts as an activator of DNA polymerases, binding to the 3′ end of primers, and promoting coordinated synthesis of both strands. Arginine and serine-rich coiled-coil 1 (RSRC1) encodes a member of the serine and arginine-rich–related protein family. The encoded protein plays a role in both constitutive and alternative pre-mRNA splicing and transcriptional regulation.
The diagnostic abilities of the key genes were examined using ROC curves (Fig. 4). The AUC values for DDX52, ESF1, HNRNPA3, MYO5A, RFC1, and RSRC1 indicated their significantly high performance (AUC value ≥70%) in discriminating IPAH patients from healthy controls, and therefore these genes were accepted as diagnostic and considered as drug targets.

ROC curves of the key genes. ROC, receiver operating characteristic.
Potential drugs identified by drug repositioning
A drug repurposing methodology was applied using CMAP data to predict drugs targeting the proteins encoded by DDX52, ESF1, HNRNPA3, MYO5A, RFC1, and RSRC1. Because these genes were upregulated in the presence of IPAH, drugs that downregulate their expression were investigated. After eliminating duplicated and unknown drugs, a total of 13 potential drugs with q-value <0.05 were found to downregulate DDX52, ESF1, HNRNPA3, MYO5A, and RFC1 (Table 1). No drug candidates were identified that downregulated RSRC1.
Potential Drug Candidates Targeting the Key Genes Associated with Pulmonary Arterial Hypertension
DDX52, DEAD-box helicase 52; HNRNPA3, heterogeneous nuclear ribonucleoprotein A3; RFC1, replication factor C subunit 1.
Discussion
IPAH is a rare but serious disease associated with significant morbidity and mortality. The pathogenesis of the disease still remains unclear. Moreover, the lack of robust molecular signatures is a barrier to therapeutic target and diagnostic discovery and development (Boucherat et al., 2017).
In this study, integrative multi-omics analyses of genome-wide expression data were performed to elucidate the underlying mechanisms of IPAH pathogenesis. The molecular profiling of IPAH was evaluated at the gene and miRNA expression levels by identifying DEGs, DEmiRNAs, and the associated molecular mechanisms. Evaluation of regulatory associations between DEGs and DEmiRNAs identified miRNAs that could be further developed as potential prognostic biomarkers.
In addition, a weighted gene coexpression network was constructed and linked to the human PPI network to identify key genes of IPAH using a network-based methodology. Modularity analysis of the constructed networks revealed the presence of common hub proteins, which were used to perform a comprehensive drug repositioning analysis. Several drugs including senolytics, alkaloids, cardiac glycosides, insulin sensitizers, and drugs with antifibrotic, anti-inflammatory, and antiproliferative effects, were suggested as potential drug candidates for IPAH.
Individual analysis of the gene expression dataset GSE15197 revealed numerous, thousands, of DEGs. However, to uncover the mechanisms behind IPAH, I combined information from gene and miRNA expression datasets, and evaluated the disease by elucidating the regulatory associations between the identified DEGs and DEmiRNAs. Overrepresentation analyses revealed significant molecular pathways and BPs. Several cancer and cancer-related pathways were found to be significantly upregulated. PAH has previously been reported to share common pathological features with cancer (Zeng et al., 2021). PAH and cancer cells show similarities in their exposure to stressful conditions, and the acquired stress tolerance of the cells allows them to survive and proliferate over time (Boucherat et al., 2017).
In addition to excessive proliferation, immune cell infiltration is a common feature of cancer and PAH (Cool et al., 2020). As expected, pathways and processes related to cardiovascular disease (CVD), lipid metabolism, and the endocrine system were also found to be significantly associated with IPAH. Lipid metabolism, particularly oxidized lipids, has been shown to contribute to many pathways involved in the pathogenesis of PAH, including vascular remodeling, mitochondrial dysfunction, proliferation and resistance to apoptosis, vasoconstriction, and endothelial dysfunction (Sharma et al., 2016). Insulin resistance in PAH patients has also been characterized by alterations in lipid metabolism and the high-density lipoprotein cholesterol fraction (Hemnes et al., 2019; Jonas and Kopeć, 2019).
The gene coexpression network reconstructed using GSE15197 IPAH data revealed two key modules via WGCNA and module–trait relationship analyses. To increase the reliability and robustness of the results, a combined intramodular connectivity was considered in the WGCNA key modules and PPI networks reconstructed around the proteins encoded by module genes. A total of 38 genes were identified as common hubs, and cross-validation analysis, followed by diagnostic performance analysis that established six key genes (DDX52, ESF1, HNRNPA3, MYO5A, RFC1, and RSRC1) as the final candidate gene signatures.
MYO5A, which is one of the three myosin V heavy-chain genes, has previously been shown to be associated with PAH. Smooth muscle myosin has been implicated in vasoconstriction in PAH, and a smooth muscle myosin inhibitor has been reported to relax pulmonary artery vascular rings in both acute and chronic rat models of PAH (Ho et al., 2012). Although not previously associated with PAH, DDX52, ESF1, HNRNPA3, RFC1, and RSRC1 are involved in a various CVDs, vascular remodeling, and proliferation.
Bioinformatic analysis identified DDX52 as a characteristic gene in heart failure (Li et al., 2022). In addition, two members of the DEAD-box family of RNA helicases, DDX46 and DDX5, have been reported to be associated with PAH and vascular remodeling, respectively. DDX46 was found to be significantly upregulated in chickens with PAH (Li et al., 2021a), and DDX5 exerted a protective effect against smooth muscle cell proliferation and vascular remodeling (Fan et al., 2019). A meta-analysis of PAH-associated transcriptomic data revealed ESF1 as commonly upregulated (Li et al., 2020). Although the role of HNRNPA3 remains unclear, increased activity of protein arginine methyltransferases, which catalyze arginine methylation of most of the heterogeneous nuclear ribonucleoproteins, has been linked to several CVDs (Couto e Silva et al., 2020; Wu et al., 2021).
RFC1 is a subunit of the replication factor C family, which may play a pivotal role in cancer cell proliferation, progression, invasion, and metastasis (Li et al., 2018). RSRC1 interacts with the transcription factor ERB2, which plays an important role in CVD (Perez et al., 2018). To the best of my knowledge, this study is the first to propose the association of DDX52, ESF1, HNRNPA3, RFC1, and RSRC1 with PAH.
Dysregulation of miRNAs contributes to the development and severity of PAH, and therefore miRNA targets play an important role in developing therapeutic strategies to reverse vascular remodeling and proliferation (Carregal-Romero et al., 2020; Grant et al., 2013). From a holistic perspective, a number of active regulatory mechanisms were identified, and represented by interactions between key genes and DEmiRNAs (Supplementary Fig. S3). Considering these associations, seven DEmiRNAs, including miR-495-5p, miR-500a-3p, miR-198, miR-517a-3p, miR-302f, miR-375 and miR-31-5p, were prominent.
Inhibition of miR-495 was reported to improve hemodynamics, vascular remodeling, and angiogenesis in PAH model (Fu et al., 2019). Although miR-500a-3p was not found to be associated with PAH, it was shown to be involved in several types of cancer (Esposti et al., 2017; Jiang et al., 2017; Long et al., 2022). miR-198 was found to play a role in neural development, cell cycle regulation, proliferation, invasion, migration, apoptosis and drug resistance, and alterations in its expression were associated with various cancers (Kaushik and Kumar, 2022; Wang et al., 2022). miR-517a-3p contributed to cell proliferation, migration, and invasion in lung cancer (Jin et al., 2014). The miR-302 family was reported to be involved in cardiomyocyte function, heart development, and regeneration in mice (Tian et al., 2015), and was also found to induce regeneration in lung tissue of Streptococcus pneumoniae–infected mice (Wang et al., 2019a).
Altered expression of miR-375 was observed in three subtypes of lung cancer, including adenocarcinoma, small cell lung carcinoma, and squamous cell carcinoma (Jin et al., 2015). In addition, miR-375 was identified as a smoking-induced miRNA in patients with chronic obstructive pulmonary disease (van Nijnatten et al., 2022). miR-375-3p was upregulated in maternal serum of women with fetal congenital heart disease (Li et al., 2021b). miR-31-5p promotes cell migration in several cancers. In addition, increased expression of miR-31-5p was found to contribute to oxidative stress and vascular smooth muscle cell migration in spontaneously hypertensive rats (Zhou et al., 2021).
These associations suggest that the resulting regulatory interactions may represent the changes in both vascular remodeling and proliferation during PAH pathogenesis and therefore, the identified miRNAs may be evaluated as potential markers for screening and therapeutic purposes in PAH.
Key gene-based drug repositioning identified alvespimycin, tanespimycin, geldanamycin, LY294002, cephaeline, digoxigenin, lanatoside C, helveticoside, trichostatin A, phenoxybenzamine, genistein, pioglitazone, and rosiglitazone as drugs with the potential to reverse gene expression (Supplementary Table S2).
Alvespimycin, tanespimycin, and geldanamycin are HSP90 inhibitors. Tanespimycin treatment has been shown to prevent the progression of PAH in a rat model, possibly by reducing inflammation through HSP90 inhibition (Wang et al., 2016). HSP90 inhibitors have senolytic activity, and senolytic drugs may hold promise for the treatment of age-related diseases or conditions associated with the burden of senescent cells (Fuhrmann-Stroissnigg et al., 2017). Mitochondrial HSP90 accumulation has been reported to contribute to vascular remodeling in PAH, and in vivo experiments showed that gamitrinib, a mitochondria-targeted HSP90 inhibitor, ameliorated PAH by reversing pulmonary vascular remodeling in two rat models (Boucherat et al., 2018).
LY294002 was a common drug found for DDX52, MYO5A, and RFC1. It acts as an mTOR, PI3K, DNA-dependent protein kinase, phosphodiesterase, and PLK inhibitor. Inhibition of PI3K/Akt signaling by LY294002 in a rat model of PAH resulted in suppression of medial smooth muscle cell proliferation but had no effect on pulmonary artery pressure, possibly because of dose (Garat et al., 2013). LY294002 was shown to reduce proliferation and chemotaxis of human pulmonary arterial smooth muscle cells in a concentration-dependent manner (Berghausen et al., 2021).
Cephaeline, the major alkaloid of ipecac, modulates G-quadruplex (G4)-dependent alternative splicing (Zhang et al., 2019). G4 formation plays a role in vital cellular functions such as transcription, translation, telomere elongation, and maintenance of genome stability. G4 structures have been proposed as promising therapeutic targets in several cancers (Varshney et al., 2020). In addition, emetine, an analog of cephaeline, was found to inhibit pulmonary artery smooth muscle cell proliferation, and ameliorate PAH in rat models (Siddique et al., 2019).
Digoxigenin, lanatoside C, and helveticoside are cardiac glycosides. Cardiac glycosides increase the force of cardiac contractions by binding to estrogen receptors or inhibiting Na+/K+ ATPase (Chao et al., 2017; Kim et al., 2015). Lanatoside C has been demonstrated to induce apoptosis of human glioblastoma and hepatocellular carcinoma cells (Badr et al., 2011; Chao et al., 2017). Helveticoside has been shown to be cytotoxic to human cancer cell lines (Kim et al., 2015; Lee et al., 2013). Given their ability to increase cardiac output, and inhibit malignant cell proliferation, the identified cardiac glycosides may have a potential role as therapeutic agents against PAH.
Trichostatin A, a histone deacetylase (HDCA) inhibitor, was a common drug found for DDX52, ESF1, and RFC1. Given the contribution of HDCA activity to vascular remodeling in PAH, Zhao et al. investigated the efficacy of two HDCA inhibitors, valproic acid, and suberoylanilide hydroxamic acid, and found that they reverse total histone levels in PAH models (Zhao et al., 2012). Trichostatin A was found to have antifibrotic and anti-inflammatory effects in adult mouse cardiac fibroblasts, supporting a potential role as a therapeutic agent to reverse remodeling in inflammatory heart disease (Somanna et al., 2016).
Phenoxybenzamine, an adrenergic receptor antagonist, is used to treat pheochromocytoma and episodes of hypertension and sweating. It has also been shown to have antiproliferative effects on human tumor cell cultures (Inchiosa, 2018). One of the tyrosine kinase inhibitors, genistein, is a natural phytoestrogen derived from soy. The protective effect of estrogen against CVD has already been demonstrated (Iorga et al., 2017; Murphy, 2011). Genistein therapy was shown to attenuate PAH and reverse cardiopulmonary function and structure abnormalities in rats (Matori et al., 2012). In addition, genistein inhibited hypertrophy of hypoxia-treated pulmonary artery smooth muscle cells isolated from chick embryos (Zhang et al., 2018b).
Pioglitazone and rosiglitazone are insulin sensitizers and PPAR receptor agonists. In a rat model of PAH, oral pioglitazone has been demonstrated to prevent right ventricular heart failure and ameliorate PAH by modulating miRNAs involved in fatty acid metabolism (Legchenko et al., 2018). Rosiglitazone treatment has also been shown to reduce pulmonary hypertension in chronically hypoxic rats (Kim et al., 2010). Considering the potential association between insulin resistance and PAH (Hansmann et al., 2007), these two insulin-sensitizing agents warrant further consideration in future clinical drug development and IPAH translational research.
This study identified a total of 13 candidate drugs including senolytics, alkaloids, cardiac glycosides, insulin sensitizers, and drugs with antifibrotic, anti-inflammatory, and antiproliferative effects, for consideration in future clinical drug development. Despite the significant results obtained here, further in vitro and in vivo studies should be conducted to evaluate the performance of the proposed repositioned drugs.
Conclusions
IPAH is a progressive and debilitating disease that affects the pulmonary arteries, resulting in increased pulmonary vascular resistance and right ventricular dysfunction, and associated with marked morbidity and mortality (Schlueter et al., 2020; Swaminathan et al., 2020). Elucidating the molecular mechanisms underlying IPAH pathogenesis is essential for developing novel systems biomarkers, diagnostics, and therapeutics. In this study, comparative analysis of mRNA and miRNA expression data revealed active regulatory associations in disease pathogenesis, and deciphered disease-specific gene and miRNA signatures. The study identified DDX52, ESF1, HNRNPA3, MYO5A, RFC1, and RSRC1 as key gene signatures associated with IPAH. The transcriptional reprogramming of these genes was found to be regulated by seven DEmiRNAs (miR-495-5p, miR-500a-3p, miR-198, miR-517a-3p, miR-302f, miR-375, and miR-31-5p), which are noteworthy as potential novel biomarker candidates.
In addition, key gene-based drug repositioning analysis identified alvespimycin, tanespimycin, geldanamycin, LY294002, cephaeline, digoxigenin, lanatoside C, helveticoside, trichostatin A, phenoxybenzamine, genistein, pioglitazone, and rosiglitazone as potential drug candidates for future clinical drug development for IPAH. Although this study provides molecular signatures and signature-based repositioned drug candidates through multi-omics data integration, future studies are needed to evaluate their clinical value as diagnostics and therapeutics for IPAH.
Footnotes
Authors' Contributions
C.K. conceived the study, carried out the analyses, interpreted the results, and wrote the article.
Author Disclosure Statement
The author declares there are no conflicting financial interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.