
Abstract
Circular RNAs (circRNAs) are of relevance to regenerative medicine and play crucial roles in post-transcriptional and translational regulation of biological processes. circRNAs are a class of RNA molecules that are formed through a unique splicing process, resulting in a covalently closed-loop structure. Recent advancements in RNA sequencing technologies and specialized computational tools have facilitated the identification and functional characterization of circRNAs. These molecules are known to exhibit stability, developmental regulation, and specific expression patterns in different tissues and cell types across various organisms. However, our understanding of circRNA expression and putative function in model organisms for regeneration is limited. In this context, this study reports, for the first time, on the repertoire of circRNAs in axolotl, a widely used model organism for regeneration. We generated RNA-seq data from intact limb, wound, and blastema tissues of axolotl during limb regeneration. The analysis revealed the presence of 35,956 putative axolotl circRNAs, among which 5331 unique circRNAs exhibited orthology with human circRNAs. In silico data analysis underlined the potential roles of axolotl circRNAs in cell cycle, cell death, and cell senescence-related pathways during limb regeneration, suggesting the participation of circRNAs in regulation of diverse functions pertinent to regenerative medicine. These new observations help advance our understanding of the dynamic landscape of axolotl circRNAs, and by extension, inform future regenerative medicine research and innovation that harness this model organism.
Introduction
Circular RNAs (CircRNAs) are stable, conserved across species, abundantly expressed, tissue- and developmental stage specific, and one of the recently discovered RNA types. They were initially considered nonfunctional aberrant RNAs that arise from errors during the gene transcription process. However, studies have shown that circRNAs are essential to control many biological processes by acting as miRNA sponges, gene expression regulators, and interactors of proteins. Regarding their origin, circRNAs are divided into three main groups: exonic circRNAs, circular intronic RNAs, and exonic–intronic circular RNAs (Memczak et al., 2013). They lack the 5′ cap or 3′ poly-A tail and are circularized by covalent bonds (Li et al., 2018).
circRNAs play diverse biological roles in homeostasis and disease pathology through several mechanisms, mainly competing with host genes for splicing machinery, serving miRNA-binding sites, and modulating gene expression at the post-transcriptional level (Kulcheski et al., 2016). For example, ciRS-7 circRNA, also called CDR1as, functions as a typical miRNA sponge and has more than 70 binding sites for miR-7 (Hansen et al., 2013a). The inability of miR-7 to regulate its target mRNAs due to binding to ciRS-7 is observed in many cancers such as lung cancer and astrocytoma (Hansen et al., 2013b; Weng et al., 2017). In addition to being miRNA sponges, circRNAs can act as sponges for RNA-binding proteins (RBPs) to adjust protein levels. Considering the roles that RBPs take in almost all cellular processes, including proliferation, apoptosis, and differentiation, circRNAs have the potential to control almost all biological processes. For example, circMBL, a conserved among invertebrates and vertebrates, can bind to the Mannan binding lectin (MBL) protein from many sites to regulate the various pathways in muscle and nervous tissues (Ashwal-Fluss et al., 2014; Pamudurti et al., 2022).
As another example, the roles of circFOXO3 in the cell cycle and apoptosis are well described. It has been documented that the circRNA of circFOXO3 interacts with p21 and cyclin-dependent kinase 2 to form a ternary complex, which decreases the activity of cell cycle essential genes, Cyclin A and Cyclin E, and thus blocks the cell cycle progression (Du et al., 2016).
The association of aberrant circRNA expression with many diseases further supports their crucial roles in maintaining homeostasis and health. However, circRNA's functions in regeneration, particularly limb regeneration, are under investigated. Salamander axolotl (Ambystoma mexicanum) is an astonishing model organism to explore the molecular basis of organ and extremity renewal due to its exceptional functional restoration capacity (Bölük et al., 2022). Cutting-edge next-generation sequencing technologies have been adopted to dissect the alteration in gene expression profile throughout the regeneration process. Axolotl reference datasets generated from RNA sequencing, proteomics, microbiome, and metabolome analyses have expanded the current understanding of the regeneration concept (Demircan et al., 2020; Demircan et al., 2019; Demircan et al., 2017; Gerber et al., 2018; King and Yin, 2016; Liu et al., 2022; Qin et al., 2021; Rao et al., 2014; Sibai et al., 2019; Wei et al., 2022). Yet, the circRNA data for axolotl are still unavailable. To identify the axolotl's circRNAs and predict their roles in limb regeneration, the circRNA profile during the early phase of limb regeneration has been mapped.
Differentially expressed (DE) circRNAs at 1- and 14-day postamputation (DPA) compared with intact limb were listed to enrich gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome pathways. The findings will contribute toward an extension of circRNA roles and better utilization of axolotl in regeneration studies.
Materials and Methods
Experimental design and animal experiments
The Istanbul Medipol University local Ethics Committee approved all performed animal experiments with authorization number 38828770-E15956. A total of 27 randomly selected sibling wild-type axolotls (1-year-old, 12 cm in length) were used in this study to form 0-, 1-, and 14-DPA time points. Axolotls were obtained from the Istanbul Medipol University Medical Research Center and bred as described elsewhere (Demircan et al., 2017). To have repeatable accuracy, nine animals of each time point group were subgrouped into three biological replicates (R1, R2, and R3). Before amputations and sample collection, animals were anesthetized using 0.1% ethyl 3-aminobenzoate methanesulfonate (MS-222; Sigma-Aldrich, St Louis, MO, USA). Following the amputation, samples were collected at 0-, 1-, and 14-DPA around the cut site and frozen immediately to store at −80°C until RNA isolation.
RNA isolation and circRNA sequencing
TRIzol reagent (Cat. No. 15596018; Invitrogen™) was used to isolate the total RNA from frozen tissues collected at 0-, 1-, and 14-DPA according to the manufacturer's procedure. Previously established protocols (Panda et al., 2017; Xiao and Wilusz, 2019) were followed to purify circRNA from total RNA by depleting the mRNA and rRNAs. Briefly, 20 μg of total RNA samples were mixed with 20 U RiboLock RNase inhibitor (Thermo Fisher Scientific), 1 × RNase R buffer, and 20 U RNase R (RNR07250; Epicenter) to form a 20 μL reaction volume, which was incubated at 37°C for 30 min to eliminate the linear RNA. Following the phenol:chloroform sodium acetate precipitation, a poly(A) tail was added to the residual linear RNAs using a commercially available Polyadenylation Kit (AM1350; Thermo Fisher Scientific) according to the producer's protocol. The Poly(A) Purist™ MAG Kit (AM1922) was used to select the RNAs with a poly(A) tail to discard any remaining linear RNAs.
Since there is no free hydroxyl group at the 3′ end of circRNAs, polyA tails cannot be added to them, and they were not affected by this step. Purifying circRNAs was followed by preparing a sequencing library using the TruSeq Small RNA Library Prep Kit protocol according to the manufacturer's instructions (Cat No: RS-200-0012; Illumina). miRNA libraries were quantified by quantitative polymerase chain reaction (qPCR) using the KAPA Biosystems Library Quantification Kit for Illumina platforms (Cat No: KK4844; Roche). Sequencing libraries were loaded at 20 pM concentration on the Illumina NovaSeq sequencer.
Raw data processing, mapping, and quantification of circRNAs
The Trinity algorithm (Grabherr et al., 2011) was employed to perform de novo assembly of raw reads, resulting in a database of putative axolotl circRNAs. The annotations for these putative circRNAs are computed based on the best hits (lowest e-value) from BLAST+ (Camacho et al., 2009) done against 140,538 human circRNAs from Circbank (Liu et al., 2019) and CircAtlas (Wu et al., 2020) databases. The RNA-Seq by Expectation Maximization (RSEM) protocol (Li and Dewey, 2011) is implemented to compute cirRNA expression profiles based on this putative axolotl circRNA database. First, circRNA expression count vectors are computed for biological replicates at 0-, 1-, and 14-DPA time points. Then, EBSeq (Leng et al., 2013) is used to identify circRNAs DE between 0-DPA versus 1-DPA and 0-DPA versus 14-DPA time points.
Validation of circRNA sequencing results
To validate the obtained DE circRNA list by following a previously published protocol (Vromman et al., 2021), five DE circRNA were randomly selected among 0-, 1-, and 14-DPA. cDNA of rRNA and mRNA-depleted RNA samples used in library preparation were obtained employing random hexamers and the ProtoScript First-Strand cDNA Synthesis Kit (E6300S; NEB) according to the manufacturer's procedure. qPCR was carried out utilizing circRNA-specific primers (Supplementary Table S1) and the SensiFAST SYBR® No-ROX Kit (BIO98005; Bioline), following the manufacturer's suggestions. The quantitative reverse transcription-PCR (qRT-PCR) of three biological and three technical replicates was conducted using Roche LightCycler, and relative circRNA expression levels were calculated with the 2−ΔΔCt method (Livak and Schmittgen, 2001).
circRNA target prediction, enrichment analyses, and visualization
“Enhanced volcano” R package was used to display the DE circRNAs with p < 0.01 and |FC| > 2.0 on volcano plots. Lists of annotated circRNA and miRNA–circRNA interactions were downloaded from the Circbank database (Liu et al., 2019), the human circRNA database, and these two lists were merged to get a final data frame of circRNAs and corresponding miRNAs. Then, axolotl circRNA lists were matched with the merged list obtained in the previous step using the common column “CircbaseID” to identify the miRNAs potentially sponged by axolotl circRNAs. The yielded identified miRNA lists were used to find validated mRNA targets of those miRNAs by searching in “mirecords,” “mirtarbase,” and “tarbase” databases using the “multiMiR” R package (Ru et al., 2014). Found genes with EntrezID were used for GO and KEGG enrichment analyses utilizing “clusterProfiler” package in R (Yu et al., 2012). To enrich the Reactome pathways by DE circRNAs, the ReactomePA package (Yu and He, 2016) was employed. In all enrichments, to decrease the false discovery rate, p-values were adjusted with Benjamini and Hochberg procedure, and cutoffs of adjusted p-value and q-value were set to 0.05.
The “org.Hs.eg.db” (human) database was selected as the background gene list. The top enriched pathways and processes were plotted using “ggplot2” and “ggpubr” R packages.
Protein–protein interaction analyses
STRINGapp (based on STRING version 11.5) in Cytoscape was used to construct and visualize the protein–protein interaction (PPI) networks (Szklarczyk et al., 2019). To analyze the clustering and interaction of DE proteins, upregulated and downregulated proteins in 1-DPA and 14-DPA samples were queried. The top 20 DE proteins with the highest degree were picked, and the PPI network of these 20 proteins was drawn.
Results
The raw reads (221,112,171 read pairs in total, M = 24,568,019 SD = 6,476,128) generated in this study were submitted to the NCBI BioProject and SRA databases (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA852678/). The de novo assembly of raw reads resulted in the identification of 35,956 putative axolotl circRNAs, of which 34,811 had at least one orthologous human circRNA based on BLAST+ searches done against Circbank and CircAtlas databases. Principal component analysis (PCA) of the circRNA expression data (Fig. 1A) illustrated time point separation and clustering of replicates within the same group (PC1 = 55.6%, PC2 = 21.1%). PC1 demonstrated a clear separation between 14-DPA and 0-, and 1-DPA, while PC2 seemed to separate 0-DPA and 1-DPA groups. Five thousand three hundred thirty-three unique circRNAs with a human orthology identified in this study distributed across the time points as 3686 circRNA for 0-DPA, 3533 for 1-DPA, and 3286 for 14-DPA. Thirty-seven percent of these circRNAs were commonly found in all time points, whereas 23% were identified in two of the three time points (Fig. 1B).
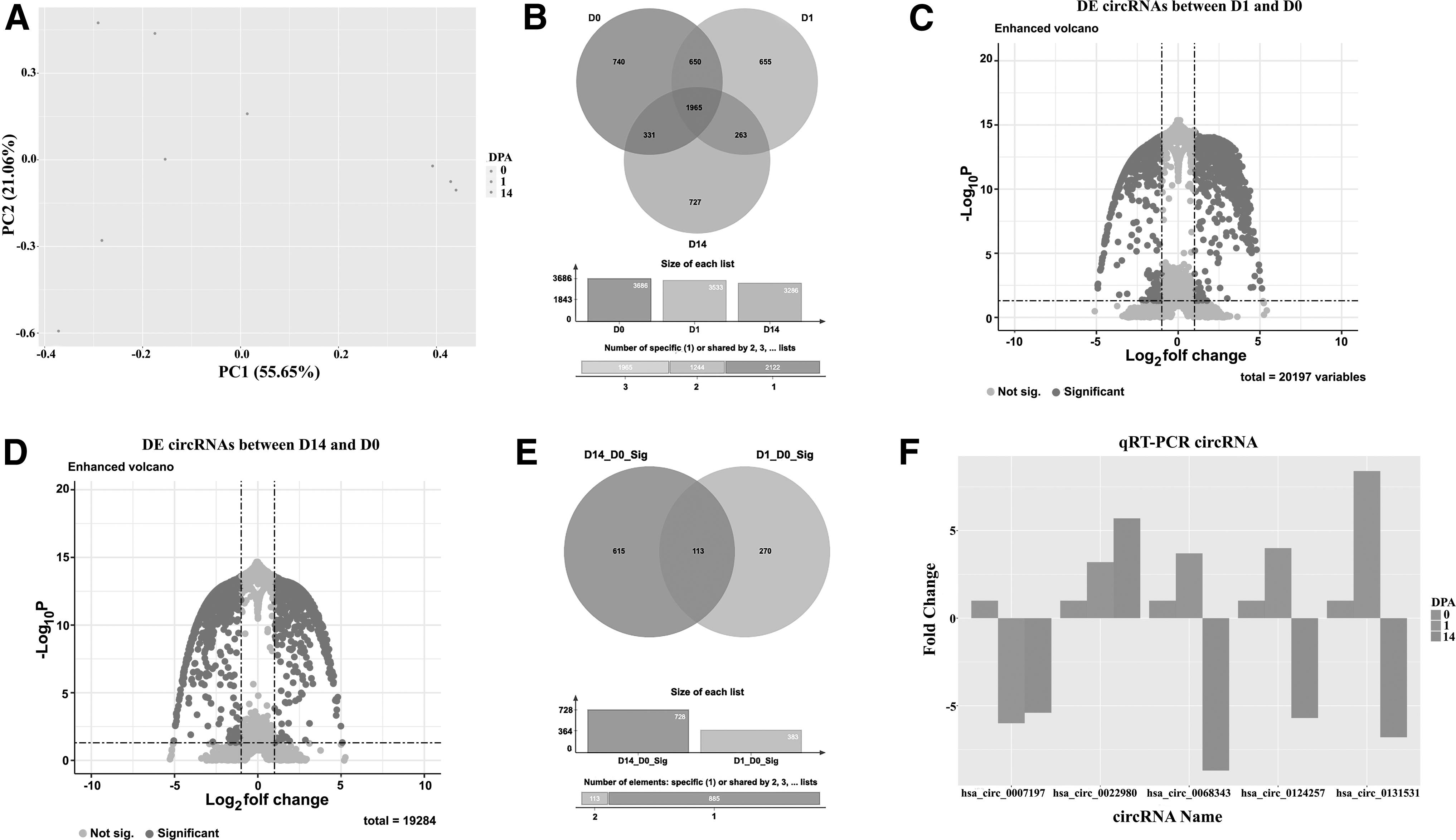
Cluster analysis was performed using circRNA expression data. PCA was generated for three time points (0-DPA, 1-DPA, and 14-DPA) and a timewise separation was observed
To analyze the RSEM data displaying log fold change against p-value, volcano plots were generated for 0- and 1-DPA, and 0- and 14-DPA comparisons (Fig. 1D, E). In the 0-DPA versus 1-DPA group, a total of 20,197 circRNAs were assessed for significance, and 323 circRNAs met the cutoff criteria (Fig. 1C, E). In contrast, for the 0-DPA versus 14-DPA comparison, out of 19,284 circRNAs analyzed, 606 circRNAs showed significance (Fig. 1D, E). One hundred forty-three circRNAs were shared between two comparisons (Fig. 1E). Validation of the sequencing and in silico analysis was performed using qRT-PCR, and the expression levels of five randomly selected DE circRNAs (hsa-circ-0007197, hsa-circ-0022980, hsa-circ-0068343, hsa-circ-0124257, and hsa-circ-0131531) in the experimental groups were measured and visualized (Fig. 1F). A similar trend between RNA seq and qRT-PCR results stimulated us to continue with downstream analysis.
Next, DE circRNAs without hits in the human circRNA database were filtered, and the miRNAs potentially interacting with the annotated DE circRNAs were investigated (Supplementary Table S2). Among the identified miRNAs, those commonly found in either upregulated or downregulated lists were excluded, and the remaining miRNAs were presented in Supplementary Table S3. At 1-DPA, 23 miRNAs were upregulated, and 79 miRNAs were downregulated, while at 14-DPA, 11 miRNAs were upregulated and 15 miRNAs showed potential downregulation as interactors of annotated DE circRNAs. Validated targets of these miRNAs (Supplementary Table S4 and Fig. 2A) were then analyzed to enrich the biological process (BP) terms, Reactome pathways, and KEGG pathways. In the comparison between 1-DPA and 0-DPA, the miRNAs upregulated and downregulated targeted 231 and 2637 genes, respectively, while in the comparison between 14-DPA and 0-DPA, the upregulated miRNAs targeted 7967 genes, and the downregulated miRNAs targeted 565 genes.
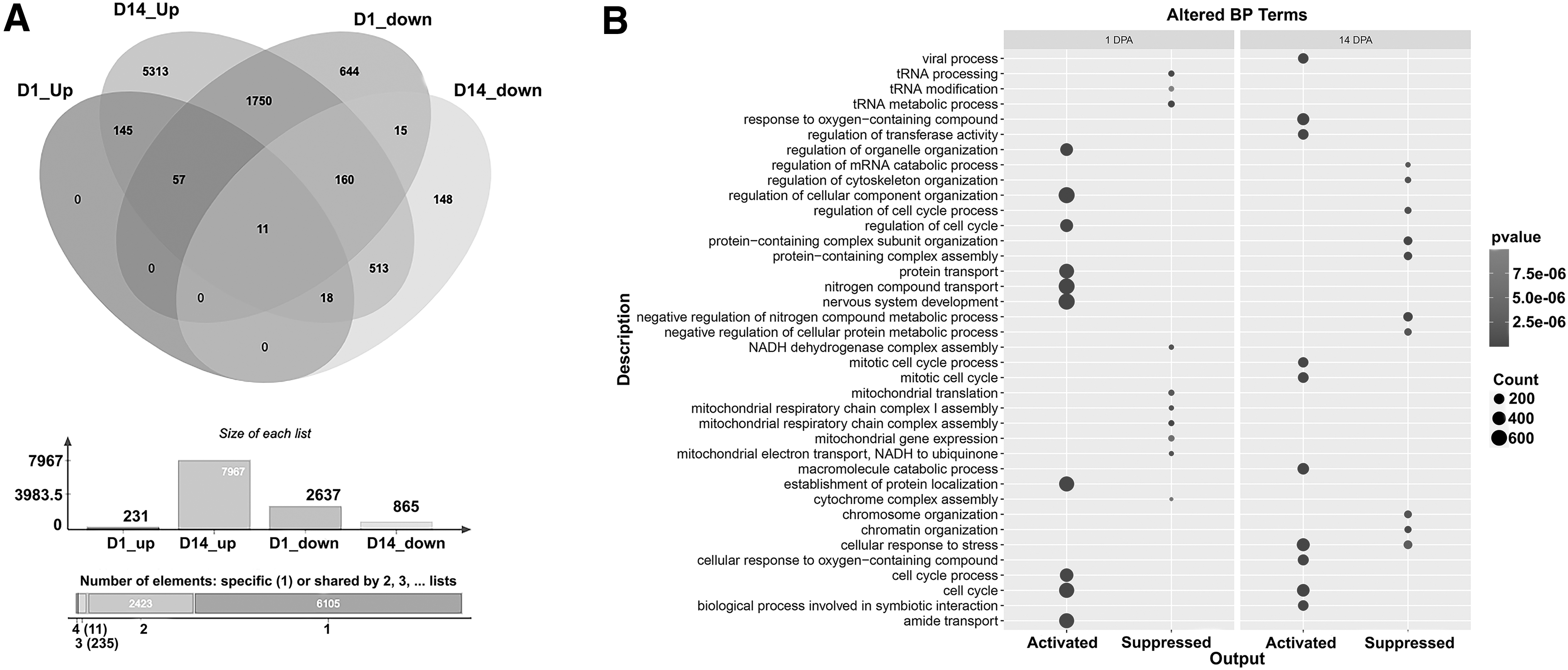
The number of genes exhibiting significant upregulation or downregulation in their expression levels on days 1-DPA and 14-DPA were depicted using a Venn diagram
The top 10 enriched BP terms in 1-DPA samples included “cell cycle,” “nervous system development,” and “nitrogen compound transport,” while the top 3 suppressed pathways were “tRNA metabolic process,” “mitochondrial respiratory chain complex assembly,” and “tRNA processing” (Fig. 2B). In contrast, the top activated BP terms in 14-DPA samples were “cellular response to stress,” “cell cycle,” and “mitotic cell cycle,” while the top suppressed terms were “negative regulation of nitrogen compound metabolic process,” “protein-containing complex subunit organization,” and “protein-containing complex assembly” (Fig. 2B). The altered BP terms are listed in Supplementary Table S5.
Furthermore, the genes enriching KEGG pathways were analyzed (Fig. 3A). The important activated pathways in the 1-DPA samples included “TGF-beta signaling pathway,” “Mineral absorption,” and “Fc gamma R-mediated phagocytosis.” On the other hand, the top 3 suppressed pathways were “Proteoglycans in cancer,” “FoxO signaling pathway,” and “Human T cell leukemia virus 1 infection.” In the 14-DPA group, the enriched pathways included “Proteoglycans in cancer,” “mammalian target of rapamycin (mTOR) signaling pathway,” “MAPK signaling pathway,” and “Autophagy—animal,” while the suppressed pathways were “Apoptosis,” “NF-kappa B signaling pathway,” and “Herpes simplex virus 1 infection.” The lists of upregulated and downregulated KEGG pathways are presented in Supplementary Table S6.

To illustrate the top 10 KEGG pathways that exhibited activation or suppression at 1-DPA or 14-DPA following an additional enrichment analysis a barplot was employed
The top 10 activated and suppressed Reactome pathways (Supplementary Table S7) at 1-DPA and 14-DPA are illustrated in Figure 3B. Notable activated Reactome pathways in 1-DPA included telomere length-related pathways, such as “Telomere C-strand (Lagging Strand) Synthesis,” “Extension of Telomeres,” and “Processive synthesis on the C-strand of the telomere,” as well as DNA repair or cell cycle-related pathways like “HDR through Homologous Recombination (HRR),” “Recruitment of NuMA to mitotic centrosomes,” and “Chromosome Maintenance.” Notch signaling-related pathways, such as “NOTCH4 Activation and Transmission of Signal to the Nucleus,” “Signaling by NOTCH4,” and “NOTCH2 Activation and Transmission of Signal to the Nucleus,” were suppressed at 1-DPA. At 14-DPA, the top activated Reactome pathways included “Mitotic Anaphase,” “Mitotic Metaphase and Anaphase,” “Semaphorin interactions,” and “Separation of Sister Chromatids.” Noteworthy Reactome pathways described at 14-DPA were “Cellular Senescence,” “DNA Damage/Telomere Stress-Induced Senescence,” “Diseases of programmed cell death,” and “Senescence-Associated Secretory Phenotype (SASP).”
Finally, the PPI network was examined for the DE proteins identified in the 1-DPA and 14-DPA samples. The top 20 DE proteins with the highest number of interactions are depicted in Figure 4. In the 1-DPA network, upregulated DE proteins, such as AKT1, MYC, and MAPK1, exhibited significant associations with downregulated DE proteins, including PTEN, TP53, and Caspase3 (Fig. 4A). Similarly, in the 14-DPA samples, upregulated DE proteins, such as TP53, MAPK3, and MYC, displayed notable interactions with top downregulated DE proteins, such as CD4, IL16, and UBC (Fig. 4B).

PPI network of top 20 DE proteins potentially regulated by identified circRNAs. PPI network was constructed between upregulated and downregulated proteins in 1-DPA
Data availability statement
All data generated or analyzed during this study are submitted to NCBI SRA databases (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA852678/) and included in supplementary information files.
Discussion
Salamanders are astonishing models due to their remarkable regenerative capacity. Compared with the mammalians' limited renewal potential, axolotl, a well-established salamander species, has been utilized to uncover the molecular and cellular program of regeneration. Despite its evident and significant contribution to extending our understanding of regeneration, the lack of several datasets, such as circRNA data, hampered its complete usage. This study aimed to profile the circRNA composition at the early phase of limb regeneration.
Most of the unique 5331 circRNAs identified in this study were not shared between all time points. Additionally, PCA of circRNA expression highlighted the clustering of samples according to collected time points. These observations may underline circRNA profile alteration during blastema formation phases. Species-specific circRNA among animals (Sun et al., 2019) and tissue-specific circRNA expression within a species (Zhou et al., 2018) were documented before. In line with our data, in a recent study, circRNAs of zebrafish blood, heart, brain, muscle, and gills were compared, and a prominent organ- and tissue-specific circRNA profile was revealed (Sharma et al., 2019). Moreover, a similar origin-specific circRNA expression pattern was detected in human organs (Xu et al., 2017). The profile of circRNAs is more dissimilar for distant organs (Xu et al., 2017) and more alike for the different regions of the same organ, as shown in the brain (Lo et al., 2020).
Although all samples were collected from the limb, the circRNA profile was found considerably distinct among samples, most probably due to the changes in cellular composition and altered biological processes for intact limb (0-DPA), wound healing phase (1-DPA), and blastema stage (14-DPA).
The expression level patterns between examined time points as observed in the RNA-sequencing results are confirmed by qPCR using stably expressed circRNAs with similar expression levels in all time points for normalization. Since there is no established method to assess the function of circRNAs directly, DE circRNAS between 1- and 14-DPA and 0-DPA were queried first to find sponged miRNAs by circRNAs, then the validated target mRNAs of sponged miRNAs.
The regulatory mechanisms of circRNAs on gene expression exhibit a wide range of diversity, and despite the established approaches, the assessment of circRNA's impact on cellular activities lacks a standardized bioinformatics method. The first approach involves linking circRNAs to their respective host genes. Through diverse mechanisms, circRNAs exert regulatory control over their host genes. They function as sponges for RBPs, thereby modulating the stability of these RBPs. Furthermore, circRNAs can interact with RNA polymerase, DNA demethylase, and DNA methyltransferase, influencing the transcriptional regulation of their originating host genes. Consequently, inferring a straightforward regulatory pattern of host genes by circRNAs poses challenges, although several reports have suggested the negative regulation of host genes by circRNAs.
In our dataset, during the initial phase of axolotl limb regeneration, we observed significant downregulation of two circRNAs, namely hsa_circTMEM39B_003 and hsa_circPIP4K2B_011. Notably, a previous study in zebrafish highlighted the involvement of TMEM39B, an endoplasmic reticulum-localized transmembrane protein, in stress resistance and tissue repair (Liu et al., 2022). Additionally, PIP4K2A, a kinase responsible for phosphorylating and removing nuclear PI5P, plays roles in gene expression changes associated with cell-cycle progression, epithelial–mesenchymal transition, reactive oxygen accumulation, and metabolism. A prior investigation underscored its negative regulation of myotube formation (Stijf-Bultsma et al., 2015). Given the crucial roles of TMEM39B in tissue repair and PIP4K2A in suppressing myogenesis, the downregulation of hsa_circTMEM39B_003 and hsa_circPIP4K2B_011 during the early phase of axolotl limb regeneration may augment the expression level and/or stability of TMEM39B and PIP4K2A mRNAs, thereby facilitating a proper regenerative process.
Furthermore, we observed the upregulation of two prominent circRNAs, hsa_circCHKA_005 and hsa_circNCOR1_010. Choline kinase A (CHKA), a key enzyme involved in phosphatidylcholine biosynthesis, is known to be suppressed by a conserved microRNA regulatory circuit during axolotl limb and zebrafish fin regeneration (King and Yin, 2016). Conversely, nuclear receptor corepressor 1 (NCOR1), a chromatin-associated protein, plays a critical role in suppressing fibrosis during axolotl limb regeneration (Rao et al., 2014). Based on existing literature, one would expect the downregulation of CHKA and the upregulation of NCOR1 during axolotl limb regeneration. However, the observed inconsistency may be attributed to the dual regulatory role of circRNAs in both negative and positive regulation of their host genes or the involvement of other gene expression regulatory factors beyond circRNAs.
On the other hand, in the 14-DPA samples, the top-upregulated circRNAs were hsa_circTANC2_010 and hsa_circCHKA_005, while hsa_circEIF4G1_007 and hsa_circPLA2G4C_006 were the top-downregulated circRNAs. TANC2, a multidomain adaptor/scaffolding protein, has a negative regulatory effect on the mTOR pathway, thereby limiting cell survival capacity (Kim et al., 2021). Notably, consistent with the observations in the 1-DPA samples, CHKA was downregulated in the 14-DPA samples. EIF4G, associated with transcriptional elongation, exhibits increased expression levels during axolotl limb regeneration (Voss et al., 2015). Additionally, PLA2G4C has been shown to play a crucial role in tissue repair, particularly in nervous system regeneration (López-Vales et al., 2008). Considering the established roles of these genes and their significance in the regeneration process, as reported in previous studies, we can deduce that the identified circRNAs in this study exert a negative regulatory influence on their respective host genes.
The second approach focuses on the role of circRNAs as miRNA sponges, preventing miRNAs from negatively regulating target mRNAs. Following these approaches, we generated lists of upregulated and downregulated mRNAs and utilized these lists to enrich GO terms, KEGG pathways, and Reactome pathways. In the 1-DPA results, we observed the upregulation of pathways related to telomere length maintenance, cilium assembly, cell cycle regulation, and nitrogen compound transport (Fig. 3A), which aligns well with findings in the literature. Telomere length maintenance is critical for preventing cellular senescence, and it has been documented that axolotls exhibit elevated telomerase activity during limb regeneration to ensure the stability of telomeres for successful renewal (Springhetti et al., 2022). Cilium assembly is initiated in response to developmental signals secreted in the wound zone following axolotl amputation or injury (Wang and Dynlacht, 2018).
Studies have demonstrated that cell cycle-related pathways are activated during limb regeneration, playing a crucial role in blastema formation and regeneration (Sibai et al., 2020; Sibai et al., 2019). Furthermore, a meta-analysis study revealed the upregulation of nitrogen compound transport in blastema tissue during axolotl limb regeneration (Sibai et al., 2019).
Conversely, at 1-DPA, we observed the downregulation of tRNA processing and tRNA modification terms, translation pathways, and Notch signaling-associated pathways. During the early phase of limb regeneration, the downregulation of tRNA processing and modification terms also affects the translation rate. Notch signaling plays a vital role in development and homeostasis. Inhibition of the Notch signaling pathway during the early stages of regeneration has been shown to enhance regeneration capacity in invertebrates and axolotls (El Bejjani and Hammarlund, 2012; Yandulskaya et al., 2022). Hence, the enrichments derived from circRNA analysis are broadly consistent with previously published studies.
In the 14-DPA samples, we observed a remarkable activation of cell cycle-related terms and pathways. During the blastema stage, cells undergo a coordinated process of cell division, which is essential for tissue renewal (Bölük et al., 2022). The accumulation of progenitor cells and their subsequent differentiation into specialized cell types are crucial for proper tissue regeneration, as extensively characterized in previous studies (McCusker et al., 2015; Stocum, 2017). Therefore, the enrichment of cell cycle-related pathways using the circRNA dataset represents a significant finding, highlighting the potential prominent contribution of circRNAs in the regeneration process.
In contrast, those associated with cellular senescence and cell death are noteworthy among the suppressed terms and pathways. Previous studies have demonstrated the elimination of present senescent cells during limb regeneration by macrophages through the axolotl's unique mechanism for sensing and clearing senescent cells (Yun et al., 2015). In line with this, it has been documented that during axolotl limb regeneration, the rate of cell death significantly decreases as a mechanism to contribute to tissue growth following damage or amputation (Wells et al., 2021).
Based on the PPI data, due to the altered levels of circRNAs at 1-DPA, activated key signaling molecules, including, AKT1, MYC, and MAPK1 interacted with suppressed PTEN, TP53, and caspase3. For 14-DPA, there was a significant interaction between upregulated TP53, MAPK3, and MYC, and downregulated CD4, IL16, and UBC. These findings provide significant insights into the regulatory role of circRNAs in the early stages of regeneration process. AKT1, a serine/threonine kinase, plays a crucial role in promoting cell survival, proliferation, and migration, thus facilitating tissue repair (Carbonell et al., 2022). The upregulation of AKT1 suggests its involvement in the activation of pro-regenerative signaling pathways. MYC, a transcription factor, is known for its involvement in cellular growth and proliferation. Its increased expression indicates its contribution to the promotion of cell proliferation during wound healing and blastema phases (Jhamb et al., 2011). MAPK1 is responsible for transmitting extracellular signals to the nucleus and is implicated in cell differentiation and tissue regeneration. Its activation indicates its participation in signaling cascades that promote tissue renewal (Wen et al., 2022).
Conversely, the observed downregulation of PTEN, a tumor suppressor protein, implies its inactivation during wound healing, potentially leading to enhanced cell survival and proliferation (Park et al., 2008).
TP53, a well-known tumor suppressor gene, plays a vital role in regulating cell cycle arrest, DNA repair, and apoptosis. Its downregulation signifies a potential inhibition of apoptotic pathways, enabling cell survival and tissue regeneration (Yun et al., 2013). Additionally, the inactivation of caspase3, a key effector in the apoptotic pathway, further supports the notion of suppressed apoptosis during regeneration (Johnson et al., 2018). CD4, a surface glycoprotein found on T helper cells, plays a critical role in immune system regulation and the activation of immune responses. The observed inactivation of CD4 represents a potential suppression of immune activation during wound healing, which may be important for maintaining an appropriate balance between proinflammatory and anti-inflammatory processes (Li et al., 2020). IL16, an interleukin involved in immune cell recruitment and modulation, is known to contribute to immune responses and inflammation. The inactivation of IL16 indicates a potential dampening of inflammatory responses during wound healing, which could be beneficial for tissue repair and resolution of the inflammatory phase (Mescher, 2017).
Collectively, these findings highlight the intricate regulatory role of circRNAs in modulating the expression and activity of critical signaling molecules involved in wound healing, providing a deeper understanding of the underlying molecular mechanisms in this complex biological process.
The study has several limitations. Human orthologs were used for the annotation and enrichment of the identified circRNAs due to the lack of comprehensive axolotl circRNA and miRNA databases. Additionally, this study lacks wet-lab validations of the identified circRNAs by modulating their expression level during regeneration.
Conclusions
To the best of our knowledge, this study represents the first comprehensive map of circRNAs in axolotl to date. In this study, we present the landscape of circRNAs in axolotl during early phase of limb regeneration. Our investigation describes a list of axolotl circRNAs and reveals that a subset of these circRNAs share orthologous genes with humans, highlighting the potential for translating the messages to human by studying the biological functions and regulatory networks of circRNAs in axolotl. Subsequent bioinformatics analysis indicated that DE circRNAs may contribute to the telomere length maintenance and positively regulate the cell cycle-related processes. Concordantly, circRNAs may suppress cell death and cellular senescence-associated pathways during this process. We anticipate that our findings can serve as a foundation for understanding the circRNAs and their regulatory networks in regeneration, using the axolotl model. These new observations also inform future regenerative medicine research and innovation that harness this model organism.
Footnotes
Authors' Contributions
T.D.: methodology, formal analysis, data curation, visualization, conceptualization, resources, and writing–original draft preparation. B.E.S.: methodology, software, and writing–review and editing. All authors have read and agreed to the final published version of the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK; Project No. 120Z217), and by the BAGEP Award of the Science Academy.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.