
Abstract
Checkpoint kinase 1 (CHK1), a serine/threonine kinase, plays a crucial role in cell cycle arrest and is a promising therapeutic target for drug development against cancers. CHK1 coordinates cell cycle checkpoints in response to DNA damage, facilitating repair of single-strand breaks, and maintains the genome integrity in response to replication stress. In this study, we employed an integrated computational and experimental approach to drug discovery and repurposing, aiming to identify a potent CHK1 inhibitor among existing drugs. An e-pharmacophore model was developed based on the three-dimensional crystal structure of the CHK1 protein in complex with CCT245737. This model, characterized by seven key molecular features, guided the screening of a library of drugs through molecular docking. The top 10% of scored ligands were further examined, with procaterol emerging as the leading candidate. Procaterol demonstrated interaction patterns with the CHK1 active site similar to CHK1 inhibitor (CCT245737), as shown by molecular dynamics analysis. Subsequent in vitro assays, including cell proliferation, colony formation, and cell cycle analysis, were conducted on gastric adenocarcinoma cells treated with procaterol, both as a monotherapy and in combination with cisplatin. Procaterol, in synergy with cisplatin, significantly inhibited cell growth, suggesting a potentiated therapeutic effect. Thus, we propose the combined application of cisplatin and procaterol as a novel potential therapeutic strategy against human gastric cancer. The findings also highlight the relevance of CHK1 kinase as a drug target for enhancing the sensitivity of cytotoxic agents in cancer.
Introduction
Cancer is a major planetary health burden. Gastric cancer is one of the most leading causes of cancer death worldwide. The early-stage gastric cancer can be managed by endoscopic resection, while suboptimal surgical resection along with chemoradiation is the treatment for locally advanced gastric adenocarcinoma (Orditura et al., 2014). Recent therapeutic advances include checkpoint inhibitors and biomarker-directed therapy, which offer prolonged survival in patients with metastatic gastric adenocarcinoma (Nevo and Ferri, 2023). Public health diagnostics innovation for prevention and precision therapeutics in cancers is one way to reduce the global burden of gastric cancer and other cancers around the world (Dzobo, 2021).
Cell cycle checkpoints are major contributors to the complex regulatory mechanisms that cause cancer, and thus considered veritable targets for public health diagnostics discovery and development. Among the checkpoints associated with cancer, kinases are the most intriguing factors linked to tumor formation and progression (Fabbro and Garcia-Echeverria, 2002). Checkpoint kinase 1 (CHK1), Pim-1 proto-oncogene serine/threonine kinase (PIM1), MAPK/extracellular signal-regulated kinase (MEK1), and cyclin-dependent kinases (CDKs) have all recently been identified as promising targets for the development of cancer progression inhibitors (Chang et al., 2013; Malumbres and Barbacid, 2009; Merkel et al., 2012; Tao and Lin, 2006).
In response to DNA damage, CHK1 initiates the phosphorylation of both ATR (ataxia telangiectasia and Rad3 related) and ATM (ataxia telangiectasia, mutant), accompanied by phosphorylation of a range of substrates and the initiation of signal cascades that culminate in cell cycle arrest (Shiloh, 2001). The checkpoint effectors (Cdc25A and Cdc25C, p53, and BRCA1) are phosphorylated/activated when ATM and ATR are activated, which is accomplished by phosphorylation/activation of CHK1 and CHK2 kinases (Sorensen et al., 2003).
CHK1 kinase overlays the core cell cycle machinery to ultimately control Cdc2 activation (Tapia-Alveal et al., 2009). It induces cell cycle arrest by facilitating the degradation of Cdc25 phosphatase at S-76 in S and G2/M phases (Wang et al., 2002). CHK1, considered an oncogene in tumors, is a DNA damage sensor and cell death pathway stimulator. Its actions are crucial for tumorigenesis and for the survival of cancer cells treated with chemotherapy and radiotherapy (Zhang and Hunter, 2014).
Given the growing importance of CHK1 inhibitors in cancer treatment, there is a demand for a wider chemical range of CHK1 inhibitors. CHK1's distinguishable role in the DNA repair mechanism of normal and tumor cells exposes it as a master regulator, making it a viable emerging biological target for therapeutic intervention. Unfortunately, there are only few clinical trial candidates in the early phases (Walton et al., 2016). However, barriers in novel drug development, such as prolonged time, large capital, and effort, encourage us to look for alternative strategies (Dugger et al., 2018).
Cisplatin is one of the most effective and widely used chemotherapeutic agent for the treatment of multiple cancer types. It is a crosslinker that damages DNA by inducing both interstrand and intrastrand crosslinks, as well as protein–DNA crosslinks (Florea and Busselberg, 2011). It is conventionally considered a cell cycle agonist as it produces heterogeneous responses, causing either cell death or cell cycle arrest, leading to the persistence of cancer cells (Galluzzi et al., 2012). The regulation of balance between cell cycle arrest and cell death in response to cisplatin treatment is poorly understood. The duration and dose of cisplatin play major roles in affecting the cell cycle phase or overall cytotoxicity in cancer cells (Luong et al., 2016). These observations suggest that combinatorial strategies of cisplatin with inhibitors of cell cycle checkpoints could potentially overcome cancer resistance.
A study reported that the inhibition of CHK1 with cisplatin induced mitotic cell death in p53-deficient small cell lung cancer (SCLC) cells, and CHK1 inhibitors such as prexasertib and AZD7762 enhanced cisplatin antitumor activity and overcame cisplatin resistance in SCLC (Hsu et al., 2019). In ovarian cancer cells, cisplatin is used in combination with other chemical agents or compounds such as honeybee venom (Alizadehnohi et al., 2012), withaferin (Kakar et al., 2012), and trichostatin A or 5-aza-2′-deoxycytidine (Meng et al., 2013) in both resistant and sensitive cell lines.
Procaterol, a selective β2-adrenoceptor agonist, is reported as an effective CDK12 inhibitor known to restrict gastric cancer cell proliferation and tumor growth (Liu et al., 2020). In cancer cells, cisplatin resistance is possibly due to the intracellular accumulation of cisplatin, which is modulated by Na+ or K+-ATPase activity. The combination of cisplatin with procaterol treatment in non-small cell lung cancer (NSCLC) cells improves sensitivity to cisplatin by enhancing Na+ or K+-ATPase activity, thereby increasing intracellular accumulation of cisplatin (Bando et al., 2000). This suggests the potential anticancer activity of procaterol.
Taken together, based on these insights, we employed in silico drug repurposing strategies, including e-Pharmacophore modeling, molecular docking, molecular dynamics (MD), molecular mechanics, and absorption, distribution, metabolism, and excretion (ADME) prediction, to identify an effective CHK1 selective inhibitor for a better and more efficient treatment regimen in gastric cancer, in particular. Furthermore, we studied the combination of procaterol and cisplatin by determining cytotoxicity, proliferation behavior, and cell cycle progression in gastric cancer to ascertain the action of procaterol on cell cycle, where CHK1 plays a key role in regulation.
Materials and Methods
Protein preparation and its structure validation
The three-dimensional (3D) co-crystallized structure of the CHK1 protein in complex with the clinical trial CHK1 inhibitor (CCT245737) was sourced from the Protein Data Bank (PDB) with the ID 5F4N and a resolution of 1.91Å. This structure was then prepared using the “Protein Preparation Wizard” from the Schrödinger suite (2019\1; Schrödinger, LLC, New York, NY, USA).
In the protein preprocessing stage, bond orders were adjusted, and coordinates for missing side chains and loops were fixed using PRIME. In addition, missing hydrogen atoms were added. All water molecules situated less than 5Å from the co-crystallized ligand were retained to facilitate protein-ligand binding interactions. Subsequently, energy minimization was performed using the OPLS3e force field. The processed and minimized CHK1 protein structure was validated by inspecting the phi/psi distribution on the Ramachandran Plot, as determined by the PROCHEK analysis. Similarly, the CHK2 protein (PDB: 2CN8) was prepared, minimized, and validated in the same manner as CHK1 for the purpose of selectivity evaluation.
Receptor grid generation
A receptor grid was generated using the OPLS-3e force field through the “Receptor Grid Generation” panel of the Schrödinger suite (2019-1; Schrödinger, LLC). By selecting the hit atoms of the co-crystallized ligand, both internal and external grid boxes were created to delineate the ATP binding pocket for inhibitors. This step ensures the removal of the co-crystallized ligand from the protein binding site, paving the way for molecular docking of the ligand library.
e-Pharmacophore hypothesis generation
In our study, we employed the PHASE v3.8 module from the Schrödinger suite (2020-2; Schrödinger, LLC) to construct an e-pharmacophore model. The criteria for database screening were derived from the superior re-docked pose of the co-crystallized ligand (CHK1 inhibitor CCT245737), with insights gained from the extra precision (XP) visualizer detailing reward and penalty regions. The phase module identifies six feature types: Hydrogen bond acceptors (A), hydrogen bond donors (D), hydrophobic groups (H), negatively ionizable groups (N), positively ionizable groups (P), and aromatic rings (R). With insights from the XP visualizer and the existing understanding of CHK1 protein-ligand interactions, we manually selected the critical chemical features for subsequent database screening.
Ligand preparation
All existing drugs were sourced as two-dimensional structure data files from the “Drugbank” database and subsequently processed using the “LigPrep” software package of GLIDE V7.7 (2019-1; Schrödinger, LLC). The structural refinement was undertaken with several key parameters: Using the OPLS-3e force field for energy minimization, generating ionization states at a pH of 7.0 ± 2.0, producing appropriate tautomers with Epik v4.0, activating the desalt option to neutralize the structure, and preserving chirality by setting the system to produce up to 32 isomers per ligand. The conformers produced in this ligand preparation stage were integrated into a database through the “Create Phase Database” panel of the PHASE v3.8 module in the Schrödinger suite. During this database creation, ligands were screened based on QikProp properties, Lipinski's rule, and the presence of reactive functional groups.
Pharmacophore-based virtual screening
The e-pharmacophore underwent a virtual database screening to identify candidates from the existing drug database that matched the pharmacophore, using the PHASE v3.8 module of the Schrödinger suite (2019-1; Schrödinger, LLC). This screening process identified ligands with chemical features analogous to those of the co-crystallized clinical trial inhibitor. Once screened, the pharmacophore-matched ligands were ranked based on a fitness score that ranged from 0 to 3, as per the criteria set in the PHASE.
Molecular docking
Before commencing molecular docking, the accuracy of the docking program was assessed by calculating the root mean square deviation (RMSD) value. This was done by extracting the co-crystallized CHK1 inhibitor from its binding pocket, re-docking it to the same active site of the CHK1 protein, and then superimposing this re-docked ligand conformation onto the original native ligand to compute the RMSD value. An RMSD value under 1 affirmed the reliability of the docking program.
The e-pharmacophore-matched existing drug candidates, which were identified from database screening, were then subjected to the molecular docking protocol using GLIDE V7.7 in the Schrödinger suite (2019-1; Schrödinger, LLC). These compounds underwent flexible docking within the ATP binding pocket of the CHK1 protein, using the XP scoring function to gauge protein-ligand binding affinity. For the top-performing 10% of docked ligands, parameters like docking score, G score, and nature of protein interactions were manually assessed with MAESTRO (Schrödinger), and the most promising drug candidates were selected for deeper computational analysis.
QikProp ADME/toxicity property prediction
Using the QikProp v5.4 module, part of the Schrödinger suite (Maestro11.9.011, 2019-1; Schrödinger, LLC), we computationally predicted the pharmacokinetic and toxicity properties of selected drug candidates, focusing on parameters that are experimentally relevant. In addition, the module enabled the assessment of drug-like attributes by applying Lipinski's rule-of-five and Jorgensen's rule-of-three violations.
Molecular mechanics-generalized born surface area calculation
The influence of solvent on the ligands' binding free energies (ΔG bind) was determined using the prime molecular mechanics-generalized born surface area (MM-GBSA) tool in the PRIME v3.5 module of the Schrödinger suite (2019-1; Schrödinger, LLC). In brief, the Glide XP-docked poses of both the co-crystallized clinical trial inhibitor and the selected existing drug candidates were minimized. Total free binding energies were then computed, using the VSGB solvent model and OPLS-3e force field, keeping other parameters consistent. The binding free energies were derived from the equation,
In this equation, ΔEMM represents the energy difference between the CHK1 protein-inhibitor complex and the combined energies of the separate CHK1 protein and inhibitor. ΔGSolv signifies the variation in GBSA solvation energies between the complex and unbound states of the protein and inhibitor. Finally, ΔGSA indicates the energy difference based on surface areas between the complexed and unbound forms.
Molecular dynamics
The MD simulations are advanced computational tools that can be utilized effectively to analyze the stability, RMSD, and interaction of selected ligands in a flexible macromolecular environment using DESMOND V5.2 module of Schrodinger suite (2020-2; Schrödinger, LLC). During simulation, the atoms of protein-ligand complex were allowed to interact for a brief period of 200 ns and the physical movements of complex were evaluated at atomistic level. First, the docked protein-ligand complex was incorporated in orthorhombic simulation box solvated with TIP3P (transferable intermolecular potential with three points) explicit water model.
The simulation system was again recalculated to neutralize the overall charge by adding suitable counterions in the system builder panel. In addition, 0.15 M NaCl salt concentrations were fixed, 20Å away from the ligand to simulate background isosmotic salt environment. The above prepared system was incorporated in workspace and subjected to run MD simulation using NPT ensemble at 300K temperature and 1.01325 bars atmospheric pressure with the assistance of OPLS_3e molecular mechanic's force field. During 200 ns of simulation time, the structure frames were recorded in the trajectory at every 100 ps and a total of 2000 frames were saved. Finally, the simulation interaction diagram tool of DESMOND V5.2 was used to sketch the MD simulation results.
In vitro anticancer activity
Cell culture conditions and chemicals
Gastric adenocarcinoma cells (AGS) were obtained from American Type Culture Collection (ATCC, Mannassas, VA) and grown under in vitro culture conditions in Dulbecco's modified Eagle's medium (DMEM) media (5% fetal bovine serum [FBS] and 1% antibiotic and antimycotic). The cells were maintained with 5% CO2, at 37°C, in a humidifier incubator. For the assays, the 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) reagent and propidium iodide were procured from HiMedia (India). DMEM media, FBS, and 100 × antibiotic/antimycotic solution were purchased from Gibco, Thermofischer Scientific. Procaterol and cisplatin were obtained from Merck and Celon laboratories, respectively.
Determination of cell cytotoxicity using MTT assay
To study the cytotoxic effect, MTT assay was performed on AGS cell lines treated with cisplatin alone and in combination with procaterol (Mosmann, 1983). Briefly, the AGS cells (5 × 103) were seeded into a 96-well plate and treated with different concentrations of cisplatin (2–10 μg/mL), procaterol (1250–20,000 nM), and its combination. After a 48-h incubation, MTT dye was added and allowed to form purple formazan crystals for 3–4 h. The crystals were dissolved in 100 μL of a 1:1 mixture of dimethyl sulfoxide and ethanol and measured using a multimode plate reader at 570 and 650 nm (Multiskan Sky, Thermo Fisher Scientific). Cell cytotoxicity was measured as percentage of cell viability compared to untreated control.
Cell migration assay
The migratory potential of AGS cells upon treatment with procaterol and cisplatin combination was studied by wound healing scratch assay or migration assay (Liang et al., 2007). Briefly, cells were seeded in a six-well plate and incubated until they formed a confluent monolayer. A scratch was made vertically with a 200 μL tip and then treated with the cisplatin (6 μg/mL), procaterol (5000 nM), and a combination of cisplatin+procaterol (6 μg/mL +5000 nM). The images were captured at 0, 8, 24, and 48 h using an inverted microscope (PrimoVert, Carl Zeiss) and the percentage of wound closure was evaluated using ImageJ software.
Cell cycle analysis by flow cytometry
To evaluate the cell cycle arrest in AGS cells, flow cytometry was used (Karthikkeyan et al., 2021). The cells treated with cisplatin (6 μg/mL), procaterol (5000 nM), and combination treatment (6 μg/mL +5000 nM) for 48 h, were trypsinized, washed with 1 × phosphate buffered saline, and then incubated for 30 min in the dark with a hypotonic lysis buffer containing RNase (100 μg/mL) and propidium iodide (2 μg/mL). Red fluorescence was measured after cells were acquired using an analytical flow cytometer (Guava EasyCyte, Merck Millipore). The percentage of DNA present at various cell cycle stages was calculated. The schematic illustration of the methodology followed for the virtual screening and in vitro validation has been represented in Figure 1.

Schematic illustration of the integrated methodology for virtual screening
Statistical analysis
The data are presented as the mean ± standard deviation for biological triplicates from each experiment. One-way analysis of variance and GraphPad Prism software were used to statistically analyze and visualize the data. Statistics were considered significant at p-value <0.05.
Data Availability
All data pertinent to the study are provided as part of the article.
Results and Discussion
Protein ligand interaction analysis
Human CHK1 is a nuclear protein distinguished by its highly conserved N-terminal kinase domain and a less conserved C-terminal region (Kim et al., 2016). Through X-ray crystallography, the human CHK1 kinase protein complexed with its endogenous agonist, ATP, and several clinical trial CHK1 inhibitors (including UCN-01, CCT245737, rabuseritib, prexasertib, and GDC-0575) reveals five critical regions:
the hinge region, the ribose binding pocket, the solvent-exposed region, the water pocket, and the polar region.
The hinge region, characterized by its conserved Cys-87 and Glu-85 amino acids, plays a pivotal role in inhibitory activity when ligands bind. A unique set of amino acid residues, such as Tyr-20, Lys-38, Glu-55, Asn-59, Phe-149, and Ser-147, populates the interior water pocket and polar region, which are occupied by three water molecules. Interactions in this region notably enhance CHK1 kinase selectivity. Meanwhile, the ribose binding pocket is enriched with hydrophilic amino acids like Glu-91, Glu-134, and Asn-135. Adjacent to this, the exposed region situates itself close to the hinge, where the presence of polar and electronegative groups bolsters ligand stability and kinase inhibition (Chen et al., 2010; Matthews et al., 2013; Reader et al., 2011) as depicted in Figure 2A. These insights offer a comprehensive understanding of CHK1's structural and functional intricacies, aiding in the identification, selection, and optimization of potential CHK1 inhibitors.

e-Pharmacophore modeling
Pharmacophore modeling is a method in drug discovery that identifies and represents the essential molecular features responsible for biological activity, particularly in the context of interactions with biological macromolecules. This technique focuses on the 3D configuration of these features to discern suitable ligands. e-Pharmacophore modeling, distinct from its traditional counterpart, integrates both the stereoelectronic attributes of the ligand and the energetics of its interactions with the protein structure (Arun et al., 2021). This advanced approach serves as a potent screening mechanism in virtual environments, leveraging the Glide XP scoring function to define protein-ligand interactions.
In this study, an e-pharmacophore hypothesis was formulated based on the 3D crystal coordinates of the CHK1 protein complexed with a potent clinical trial inhibitor, CCT245737. This decision was rooted in the inherent confidence in known complexes. When an e-pharmacophore is based on a known protein-ligand complex, especially from high-resolution X-ray crystallography, there is a heightened confidence that the interactions being mapped are accurate. The ligand in these scenarios has already been experimentally validated for its activity, and its binding mode is clearly depicted. Moreover, e-pharmacophores capture the interaction energies and the precise geometry of the ligand within the active site. Thus, these models are not solely based on structural data but are also guided by energetic considerations, enhancing their reliability.
With this foundation, the protein structure was sourced from the PDB (ID: 5F4N; Resolution: 1.91Å) and was refined using the protein preparation wizard. The integrity of this refined structure was verified using a Ramachandran plot, showcasing the stereochemical orientation of amino acids. Once the native ligand was extracted, it was re-docked into its original binding pocket using the Glide XP docking mode. The XP-Visualizer tool aided in visualizing the binding of this re-docked co-crystallized ligand at the ATP binding site, highlighting both reward and penalty regions (Fig. 2B, C).
Our e-pharmacophore model presents a comprehensive analysis of the binding modes and interaction profiles of two ligands, CCT245737 and procaterol, within the target protein's binding pocket, illustrated in Figure 3A, B. Through a combination of visual inspection and computational docking, the critical determinants of ligand binding were identified and compared. The ligand CCT245737 exhibited a robust interaction profile with the protein. Key features identified from our e-pharmacophore model included H-bond acceptors A1 and A3, represented by pink spheres. A3 was found to form a hydrogen bond with the specific amino acid residue Cys87 within the pocket, indicating a critical contribution to binding affinity.
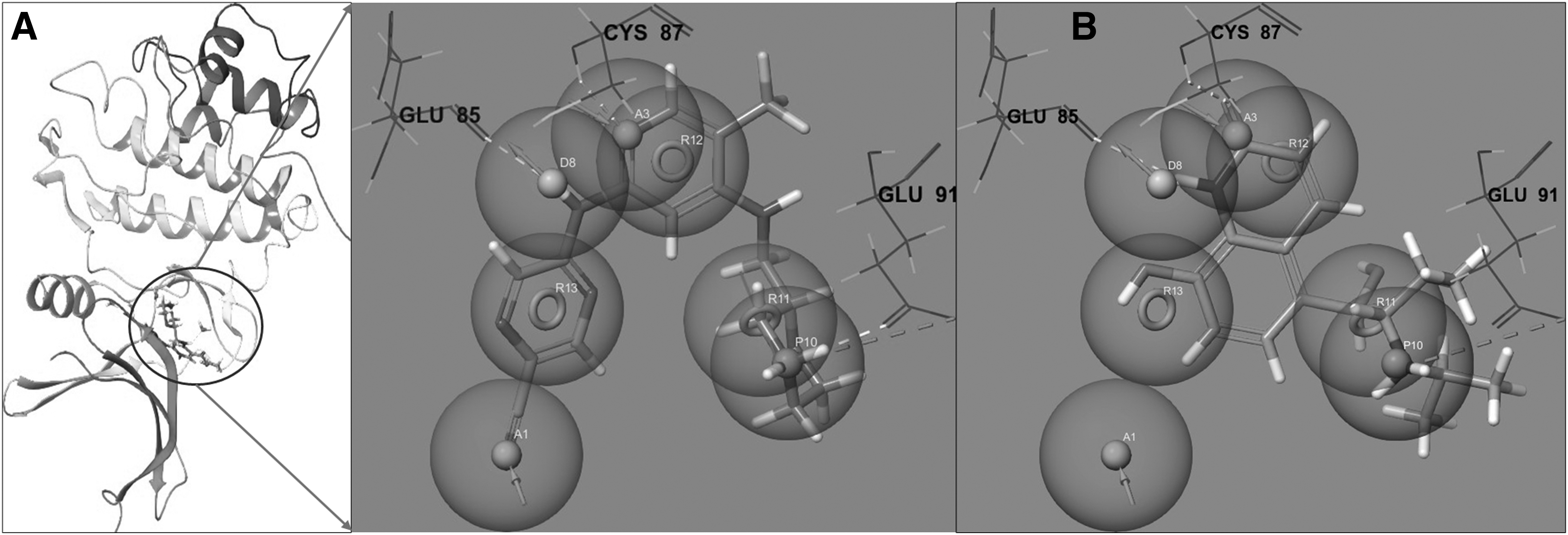
e-Pharmacophore features for database screening.
The blue sphere representing the H-bond donor feature, D8, engaged in a strong hydrogen bond with a residue Glu85, emphasizing the ligand's specificity. Another blue sphere, indicative of a positive ionizable group (P10), revealed potential ionic or hydrogen bonding interactions within the protein pocket. The aromatic rings, highlighted by the orange torus shapes (R11, R12, and R13), were deeply embedded in hydrophobic regions, suggesting their role in enhancing ligand stability within the pocket.
Procaterol, although structurally distinct, retained conserved interaction features, particularly in hydrogen bonding with Glu85 and Cys87. However, certain aromatic interactions prevalent in CCT245737 were conspicuously absent in procaterol, hinting at a differential binding mode. Critical amino acid residues, namely Glu85, Cys87, And Glu91, played pivotal roles in ligand affinity. The nature of interactions, spanning from hydrophobic to ionic and hydrogen bonds, offered insights into the ligands' binding affinity and selectivity.
A deeper dive into the chemical structures elucidated further interaction points. The presence of diverse functional groups in CCT245737, such as morpholine, pyridine, and pyrazine rings, corroborated its aromatic interactions. Conversely, procaterol's hydroxy groups emerged as potential H-bond donors or acceptors, with its propan-2-ylamino group suggesting potential polar interactions. The comparative analysis of CCT245737 and procaterol, utilizing our e-pharmacophore model, offers valuable insights into ligand-protein interactions, paving the way for optimizing ligand structures for improved binding affinity and selectivity.
e-Pharmacophore hypothesis screening and molecular docking
The pharmacophore features were incorporated into the PHASE module and were used for the previously prepared database screening. To identify a more promising existing drug for CHK1 inhibition, we set a criterion of matching a minimum of four out of the seven generated pharmacophore features. This screening resulted in 812 drug candidates, with the highest fitness score being 1.867, which were then used for further molecular docking studies.
In recent studies, molecular docking methods have been utilized to pinpoint the most suitable candidates for a given target (Luo et al., 2021). The purpose of this is to filter out drug candidates with poor binding affinity from the e-pharmacophore-based screening, leaving only the most favorable compounds. In our study, the RMSD value of the re-docked clinical trial CHK1 inhibitor (CCT245737) was remarkably low at 0.4387, confirming the accuracy of the docking program in screening and predicting the binding affinity of the aforementioned 812 ligands with the CHK1 protein (Supplementary Fig. S1).
The XP molecular docking program was subsequently employed for these existing drugs. Out of these, 167 drugs were docked into the ATP binding site of the CHK1 protein. This molecular docking yielded ligand docking scores, Glide scores, and bond interaction distance values (Ranjan et al., 2018). The binding modes of all 167 drug candidates were ranked based on these scores. The top 10% of ligands were visually analyzed to discern the nature of protein-ligand interactions and their binding affinity (Supplementary Table S1). Interaction fingerprints hinge on the specific interactions at the binding site. Structures detailing the binding modes of the top 10% ligands can be found in Supplementary Figure S2.
From the top 10% ligands, we discerned that both procaterol and dexrazoxane showcased superior binding affinity and vital interactions with the ATP binding pocket of the CHK1 protein. Specifically, procaterol exhibited two hydrogen bond interactions with the hinge region amino acid residue Glu-85 and single hydrogen bond interactions with residues Glu-91 and Cys 87 (Fig. 4A, B). Similarly, dexrazoxane formed hydrogen bonds with residues Glu-85, Cys-87, and Glu-91 (Fig. 4C, D). Both procaterol and dexrazoxane were chosen for subsequent computational analyses, based on knowledge about existing CHK1 inhibitors and the requirements of the binding site.
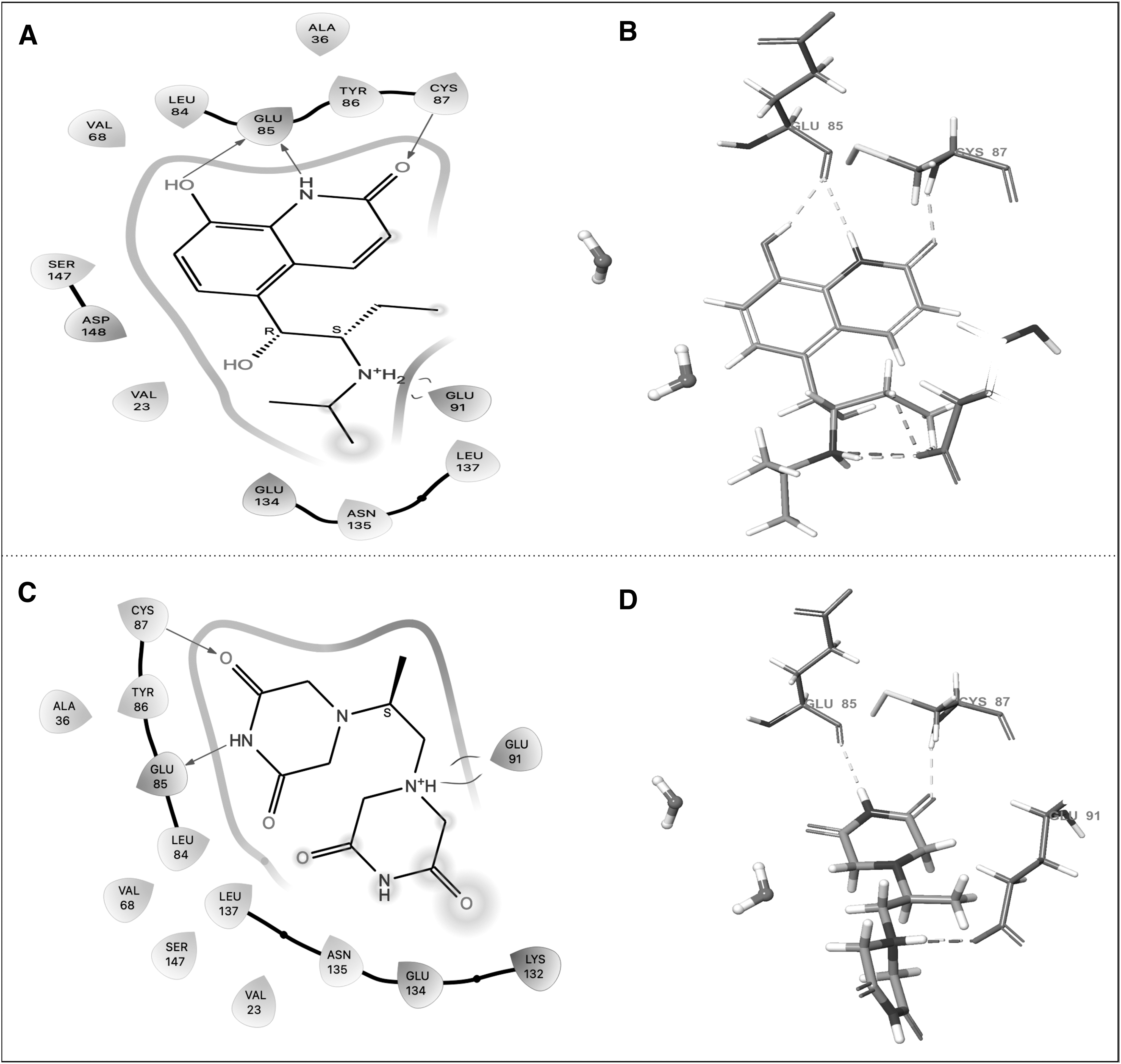
XP molecular docking results of CHK1 protein with procaterol
CHK2 kinase is a stable protein that is active throughout the cell cycle and becomes particularly active during double-strand DNA breaks, leading to cell cycle arrest at G1/S and G2/M phases and initiating apoptosis (Zannini et al., 2014). Notably, it has also been observed to induce senescence through p53 and the expression of the CDK inhibitor p21 in undamaged human cells (Antoni et al., 2007).
In this study, we further scrutinized the selectivity and potency of procaterol and dexrazoxane against the CHK2 protein using molecular docking and the Glide XP scoring function. The 3D crystal structure of the human CHK2 protein, in complex with a potent inhibitor (debromohymenialdisine), was sourced from the PDB and refined using the protein preparation wizard. Just as with the CHK1 protein, the RMSD value for superimposition was found to be modest at 0.674, confirming the accuracy of docking (Supplementary Fig. S3).
After validating the docking program, both the clinical trial chk1 inhibitor (CCT245737) and the two selected existing drugs were docked with the ATP binding pocket of the CHK2 protein using the Glide XP mode. The docking scores for CCT245737, procaterol, and dexrazoxane were −6.369, −3.497, and −5.489 kcal/mol, respectively (Supplementary Fig. S4). This molecular docking suggests that both procaterol and dexrazoxane have a lower binding affinity for CHK2 compared to the clinical trial ligand. These findings suggest that both procaterol and dexrazoxane exhibited key interactions and promising binding affinities with the ATP binding pocket of the CHK1 protein, highlighting their greater selectivity toward CHK1 compared to the CHK2 protein.
In silico prediction of pharmacokinetic (ADME) and toxicity parameters using QikProp
Generally, the QikProp module analyzes and calculates physiologically relevant pharmacokinetic and toxicity parameters, offering insights into the safety range for selected drug molecules (de Oliveira et al., 2021). In this study, we performed QikProp screening for the existing drugs, procaterol and dexrazoxane, to compare their pharmacokinetic and toxicity parameters with those of the clinical trial Chk1inhibitor. The QikProp data results are presented in Table 1. All selected drug candidates fall within the acceptable range of drug likeness characteristics.
The Selected Drug Candidates with Their Physicochemical and Toxicity Descriptors Determined by QikProp Tool
Ligand name.
MW of the compound (acceptable range: 130–725 g/mol).
Predicted octanol/water partition coefficient logP (acceptable range: −2.0 to 6.5).
Predicted aqueous solubility; S in mol/L (acceptable range: −6.5 to 0.5).
Predicted IC50 value for Blockage of HERG K+ Channels (acceptable range: below −5.0).
Predicted BBB permeability (acceptable range: −3.0 to 1.2).
Predicted apparent Caco-2 cell permeability in nm/sec (range: <25 poor, >500 great).
Number of likely metabolic reactions (range: 1 to 8).
Predicted human serum albumin binding (acceptable range: −1.5 to 1.5).
Percentage of human oral absorption (<25% is poor and >80% is high).
Number of violations in Lipinski rule of Five.
Number of violations of Jorgensen's rule of three.
BBB, blood–brain barrier; MW, molecular weight.
Prime MMGBSA calculation
The Prime MMGBSA simulation tool is employed to predict the ligand binding and strain energies of drug candidates selected from previous computational stages. The low-energy binding poses of CCT245737, procaterol, and dexrazoxane with the CHK1 protein (5F4N) from Glide XP docking were re-evaluated based on the theoretical calculations of total binding free energies. The results showed MMGBSA ΔG Bind scores for CCT245737, procaterol, and dexrazoxane to be −50.28, −48.35, and −32.40, respectively. Notably, procaterol exhibited a ΔG Bind score close to that of the clinical trial inhibitor, CCT245737, outperforming dexrazoxane. The MM-GBSA analysis thus indicates that procaterol forms a more stable complex and shows selective inhibition of the CHK1 protein, similar to the clinical trial inhibitor.
MD insight on protein-ligand stability
The MD assign velocities and calculate forces on all atoms to provide an insight into dynamic perturbations within the protein-ligand complex and interactions of ligand with water molecules. It is carried out to understand the stability and conformational changes of ligand–protein complexes. Based on the results of molecular docking, ADME/T prediction, and MMGBSA analysis, procaterol possesses better binding affinity, acceptable range of kinetic profile, and lowest ΔG Bind score than dexrazoxane. The protein-ligand stability of the above shortlisted procaterol-CHK1 complex was evaluated using Desmond MD simulation analysis and the results were compared with the reference inhibitor (CCT245737).
The MD results of the clinical trial inhibitor (CCT245737)-CHK1 complex are illustrated in Figure 5. The dynamics trajectory events remained stable throughout the simulation process. In addition, the stability of the complex was determined by plotting the RMSD graph during simulation. The Figure 5A depicts the stability of the CCT245737-CHK1 complex, and the PL-RMSD was found between the ranges of 0.50Å to 3.2Å.

The MD results of clinical trial inhibitor (CCT245737)-CHK1 complex.
Subsequently, CHK1 protein residues present in the hinge region, ribose binding pocket, and polar region interacted with the ligand atoms. The morpholine ring and adjacent amino groups in CCT245737 played a crucial role in these interactions. The complex was stabilized by different intermolecular interactions. Briefly, interaction patterns showed that 98% and 97% of H-bond interactions were formed with Glu-85 and Cys-87 in the hinge region, likely due to the pyridine nitrogen in CCT245737. Similarly, 95% of H-bond interactions was formed with the Glu-91 residue present in the ribose binding pocket, possibly due to the morpholine ring or the adjacent amino group. Also, it occupied the water pocket and forms water bridge (95% and 83%) and H-bond (39%) interactions with polar region residues Glu-55, Asn-59, and Asp-148, respectively.
The result obtained from the procaterol-CHK1 complex MD simulation is represented in Figure 6. The RMSD for the procaterol-CHK1 complex remained stable for a 200 ns timeframe, with the protein RMSD 2.25Å and ligand RMSD within 0.25Å to 4Å till the end of the simulation. Upon analyzing the specific interactions, it was found that the hydroxyl group of procaterol forms a strong H-bond interaction, 87% of the time, with the Glu-85 residue, and also establishes a water bridge interaction 87% of the time. Procaterol's carbonyl group interacts with the Cys-87 residue, resulting in a 71% H-bond interaction.
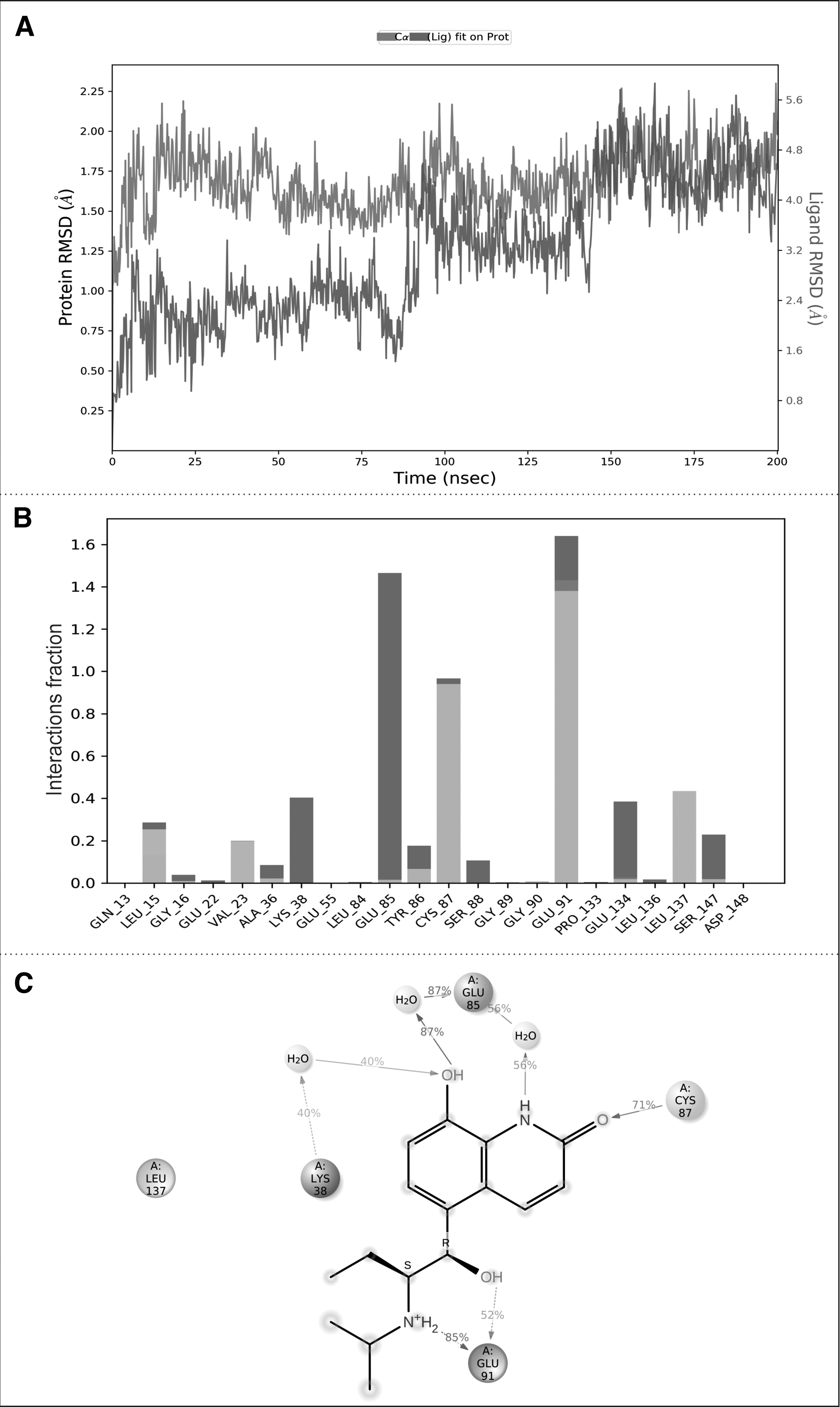
The MD results of procaterol-CHK1 complex.
Another hydroxyl group of procaterol has a water bridge interaction with Glu-91, seen 52% of the time, in the ribose binding pocket. The secondary amine of procaterol establishes an interaction with Glu-91, seen 85% of the time. Water bridge interactions, 40% of the time, are also observed with the Lys-38 and Leu-137 residues. The results of XP docking and post-MD-based intermolecular interactions for CHK1 protein with CCT245737 and procaterol are summarized in Table 2. The overall MD results insisted that procaterol is a preferable CHK1 inhibitor since it has all the essential interactions, owing to its hydroxyl groups and secondary amine, compared to the clinical trial inhibitor (CCT245737).
The Extra Precision-Docking and Post-Molecular Dynamics-Based Intermolecular Interactions Between Protein-Ligand Complexes
H bond, hydrogen bond; MD, molecular dynamics; XP, extra precision.
In summary, the interactions of procaterol with CHK1 are largely mediated by its hydroxyl, carbonyl, and secondary amine functional groups, especially in the hinge region, ribose binding pocket, and polar region.
Procaterol in combination with cisplatin significantly reduces cell viability in vitro
CHK1 is essential for proliferation, survival, and cell cycle integrity in cancer cells (Awasthi et al., 2015). Hong et al. (2012) reported that the frequent overexpression of CHK1 was correlated with poor clinical outcome in patients with hepatocellular carcinoma (HCC). They demonstrated that inhibitors of CHK1 suppress tumorigenicity through the CHK1/SYK(L) pathway in HCC in vitro and in vivo and suggested that CHK1 inhibitors may be effective as potential therapeutic agents for HCC (Hong et al., 2012). CHK1 expression was significantly higher in triple-negative ER−/PR−/HER-2-phenotype breast carcinoma and recommended that the use of CHK1 inhibitors may be a potential treatment strategy (Verlinden et al., 2007). The in silico analysis showed that procaterol is one of the existing drugs to serve as an effective CHK1 inhibitor.
Herůdková et al. (2017) reported that CHK1 inhibitor SCH900776 effectively increases the cytotoxic potential of cisplatin in human colon cancer cells (Herůdková et al., 2017). Sen et al. (2017) provided the preclinical evidence of significant efficacy of a CHK1 inhibitor as monotherapy and in combination with cisplatin in acquired cisplatin resistance small cell lung cancer (SCLC) models (Sen et al., 2017). We hypothesized that procaterol, CHK1 inhibitor, may increase the effect of cisplatin in AGS cells. To confirm this hypothesis, we performed in vitro cytotoxicity assay on AGS cells using a CHK1 inhibitor procaterol in combination with cisplatin.
AGS cells were treated with different concentrations of single drug cisplatin, procaterol, and a combination of cisplatin and procaterol. The MTT assay results showed that treatment of procaterol alone did not affect the cell viability, but in combination with cisplatin, there was significant decrease in cell viability with increase in concentration of drugs.
The IC50 values of cisplatin and procaterol combination treatment were found to be 6 μg/mL and 5000 nM/mL, respectively. Significantly, at these concentrations, there was >45% reduction in cell viability when compared to the control (Fig. 7). Previous finding by Jafri et al. (2010) showed similar results, where 60% reduction in cell viability was observed in NCI-H460 (NSCLC) cells on treatment with cisplatin alone (Jafri et al., 2010). A study by Tao et al. (2016) showed combination of cisplatin with AKT inhibitor MK-2206 drug 90% cytotoxicity on AGS cell lines (Tao et al., 2016). Hence, our study results show potential anticancer activity of procaterol in combination with cisplatin than alone.
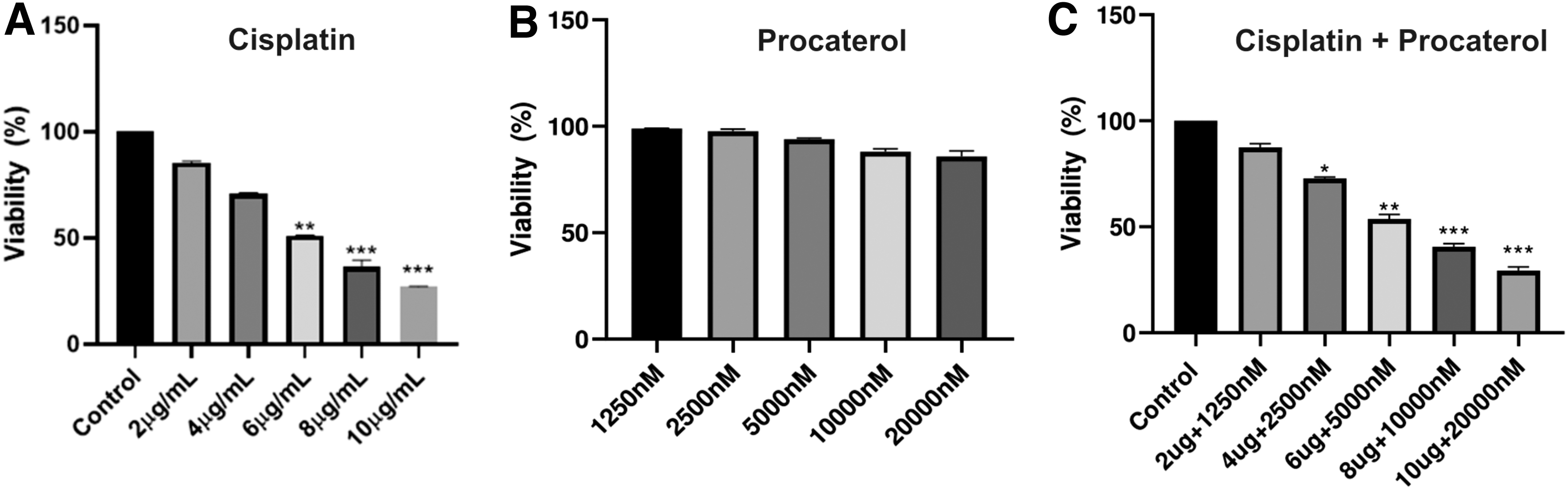
Cytotoxic effects showed by cisplatin and combination treatment on AGS cells with different concentrations of cisplatin, procaterol, and combination treatment. Cellular survival of AGS cells upon
Migration of AGS cells is inhibited in the presence of procaterol-cisplatin combination
To assess the effect of cisplatin and procaterol combination on the migration of AGS cells, wound healing scratch assay was performed. Cell monolayers were scratched and treated with cisplatin and procaterol alone and in combination. The percentage of wound closure of treated cells was evaluated at 0, 8, 24, and 48 h compared to control.
The results showed that the treatment with procaterol alone had no effect on cell migration inhibition compared to the control, while the inhibition of cell migration was observed in cisplatin alone and the migration inhibition was further enhanced in combination treatment of cisplatin and procaterol (Fig. 8). The study by Luo et al. (2019) reported that the combination of [6]-gingerol and cisplatin treatment in gastric cancer cells enhances the inhibition of migration through PI3K/AKT signaling pathway (Luo et al., 2019). Our study demonstrated that the combination of drugs showed significantly increased inhibition of cell motility compared to cisplatin and procaterol alone.
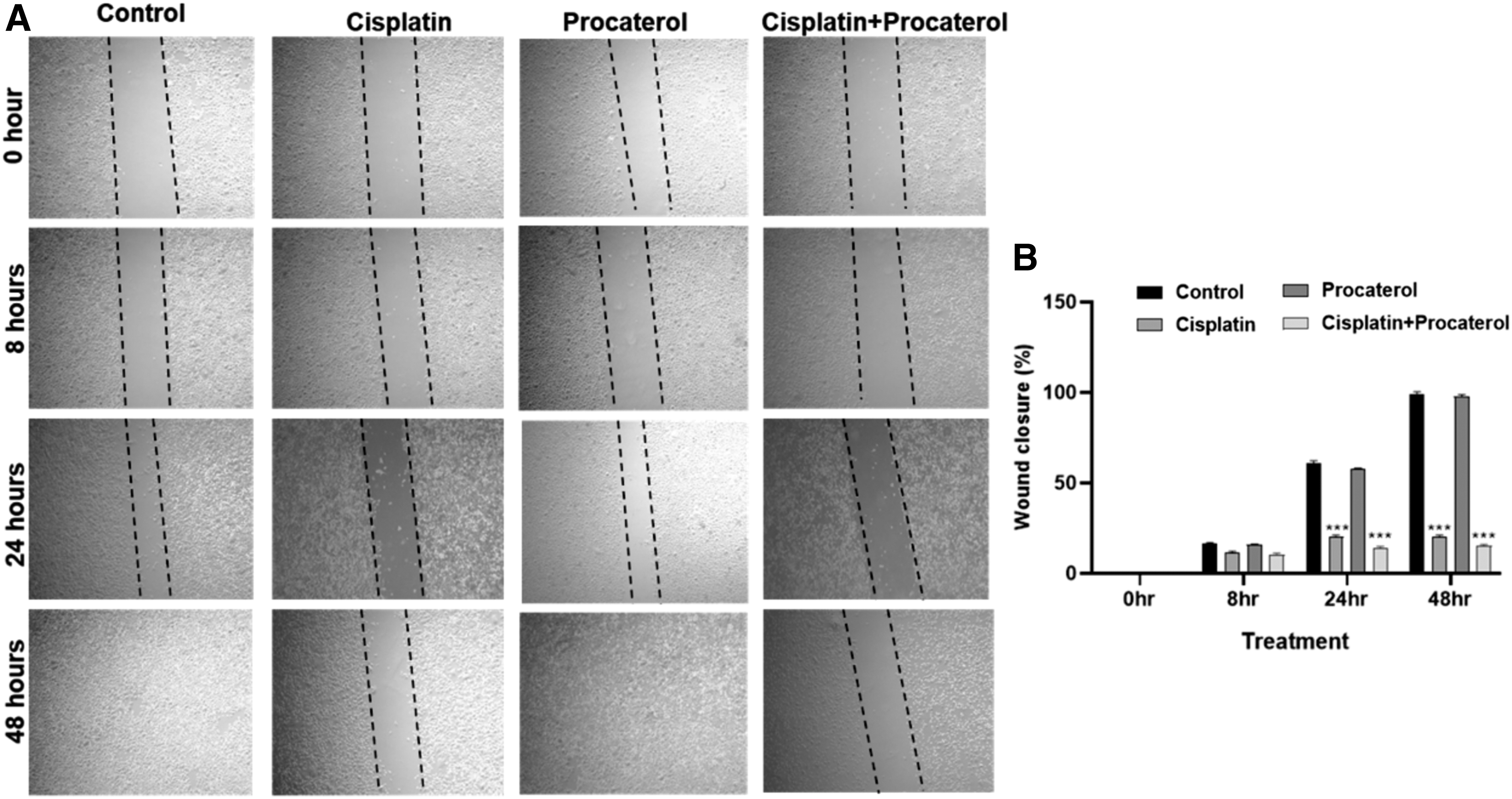
Assessment of cell cycle arrest
Cell cycle arrest is an important cause of apoptosis. Flow cytometry analysis was performed to assess the effect of cisplatin and procaterol as single drugs and combination of cisplatin and procaterol in the regulation of cell cycle. Cell cycle analysis of AGS cells after treatment with cisplatin, and in combination treatment showed cell cycle arrest at G2/M phase. The treatment with procaterol alone had no effect on cell cycle arrest compared to the control.
In comparison to cisplatin alone, the percentage of DNA content in G2/M phase is significantly higher in combination treatment (Fig. 9). A study reported that the combination therapy of cisplatin and resveratrol in gastric cancer cells induces significantly enhanced G0/G1 cell cycle arrest compared to monotherapy treatment (Rahimifard et al., 2022). This study suggested that the combination treatment of cisplatin and procaterol regulates the cell cycle at G2/M phase by significantly increasing DNA content when compared to procaterol and cisplatin treatment alone, which promotes cells to enter apoptotic or necrotic stages.

Cell cycle analysis by flow cytometry. AGS cells were treated with cisplatin, procaterol, and combination treatment.
Conclusions
This study sheds light on the promising potential of procaterol, when used in combination with cisplatin, as an effective strategy for combating gastric adenocarcinoma. The findings highlight the relevance of CHK1 kinase as a drug target for enhancing the sensitivity of cytotoxic agents in cancer. Using an integrated e-pharmacophore-based virtual screening, XP molecular docking, Prime MM-GBSA, ADMET, and MD simulation approach, we successfully identified from existing drugs a new therapeutic candidate, procaterol, which may serve as a CHK1 inhibitor. Procaterol, a long-acting β2-receptor agonist, has exhibited highly favorable characteristics, including strong binding affinity, kinetic profile, binding free energy, and complex stability within the ATP binding region of the CHK1 protein, mirroring the performance of the clinical trial inhibitor CCT245737.
The in vitro cytotoxicity assay results demonstrated a substantial reduction in cell viability when procaterol and cisplatin were administered in combination, surpassing the efficacy of their individual treatments. Furthermore, the scratch assay showed enhanced inhibition of cell migration with the combination treatment, supporting the potential synergistic effect of procaterol and cisplatin. Flow cytometry analysis revealed that the combination treatment induced cell cycle arrest at the G2/M phase, suggesting potential cell cycle regulation and promotion of cell entry into apoptotic or necrotic stages.
These findings collectively add to the body of evidence supporting the use of combination therapies in cancer treatment. Our results offer a convincing proposition that procaterol, acting as a CHK1 inhibitor, holds promise as a valuable adjuvant to augment the efficacy of cisplatin in cancer therapy, and in gastric cancer in particular, based on these findings. It is imperative that further in vivo investigations are to be conducted, coupled with an in-depth exploration of molecular mechanisms supporting the observed synergistic effects. This research identifies new molecular leads and prospects for gastric adenocarcinoma therapeutics innovation by harnessing computational and experimental approaches to cancer drug discovery.
Footnotes
Acknowledgments
We thank Yenepoya (Deemed to be University) for providing the infrastructure facility for Centre for Integrative Omics Data Science, and PSGCP and MS Ramaiah University for Schrodinger and simulation support, respectively.
Authors' Contributions
S.G.P.: Methodology, research, and article writing. J.C.: Conceptualization, methodology, visualization, and writing article. S.K.: Visualization and writing article, Editing. M.G.: Reviewing and editing. T.S.K.P.: Reviewing and editing. R.R.: Reviewing and editing. S.D.: Reviewing and editing. R.D.A.B.: Conceptualization, project administration, supervision, visualization, and writing article. All authors made a significant intellectual contribution to the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.