
Abstract
Host-virus Protein–Protein Interactions (PPIs) play pivotal roles in biological processes crucial for viral pathogenesis and by extension, inform antiviral drug discovery and therapeutics innovations. Despite efforts to develop the Epstein–Barr virus (EBV)-host PPI network, there remain significant knowledge gaps and a limited number of interacting human proteins deciphered. Furthermore, understanding the dynamics of the EBV-host PPI network in the distinct lytic and latent viral stages remains elusive. In this study, we report a comprehensive map of the EBV–human protein interactions, encompassing 1752 human and 61 EBV proteins by integrating data from the public repository HPIDB (v3.0) as well as curated high-throughput proteomic data from the literature. To address the stage-specific nature of EBV infection, we generated two detailed subset networks representing the latent and lytic stages, comprising 747 and 481 human proteins, respectively. Functional and pathway enrichment analysis of these subsets uncovered the profound impact of EBV proteins on cancer. The identification of highly connected proteins and the characterization of intrinsically disordered and cancer-related proteins provide valuable insights into potential therapeutic targets. Moreover, the exploration of drug–protein interactions revealed notable associations between hub proteins and anticancer drugs, offering novel perspectives for controlling EBV pathogenesis. This study represents, to the best of our knowledge, the first comprehensive investigation of the two distinct stages of EBV infection using high-throughput datasets. This makes a contribution to our understanding of EBV–host interactions and provides a foundation for future drug discovery and therapeutic interventions.
Introduction
Epstein–Barr Virus (EBV), also recognized as human herpesvirus 4, is a double-stranded DNA virus, belonging to the herpes virus family (Kerr, 2019). EBV was discovered in 1964 as the first human oncogenic virus in the tumor cells of Burkitt's Lymphoma (BL) (Young and Rickinson, 2004; Young et al., 2016). Implicated in a range of autoimmune disorders and malignancies in humans (Damania et al., 2022), this virus has globally disseminated its infection to nearly 95% of the adult population (Cohen et al., 2011; Epstein et al., 1964). Despite its broad planetary health importance, there remain significant knowledge gaps that hinder antiviral discovery for EBV.
EBV spreads primarily through saliva, with initial infections potentially leading to mononucleosis and resulting in a lifelong infection. This viral perseverance involves a dynamic interplay between latent and lytic modes of infection (McKenzie and El-Guindy, 2015). Latency occurs mainly in B-lymphocytes, while lytic infection can manifest in B cells or epithelial cells (Hatton et al., 2014). Globally, the EBV has significance due to its oncogenic properties and its association with multiple human malignancies, including BL, nasopharyngeal carcinoma, Hodgkin's lymphoma, gastric carcinoma, and posttransplant lymphoproliferative disease (Khan and Hashim, 2014).
The genome of EBV consists of double-stranded, linear DNA with an approximate length of 172 kb (Young et al., 2007). It encodes around 89 proteins, of which 43 are common to herpesviruses, termed core genes responsible for viral genome replication, packaging, and viral delivery in cells. The remaining 46 genes fall under the noncore gene category, of which 28 are EBV-specific, 12 are specific to the gamma subfamily and 6 have orthologs in beta and gamma herpesviruses (Kang and Kieff, 2015; Piccaluga et al., 2018; Zhang et al., 2017).
The EBV proteins involved in the latent stage are the EBV nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP) and the latent membrane proteins (LMP1, LMP2A, and LMP2B). Among these latent gene products, EBV latent proteins EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and LMP1 are essential for B-cell transformation (Zhang et al., 2017; Zhu et al., 2016). Viral genes specifically expressed during the productive replication cycle are termed “EBV lytic genes” (Kalla et al., 2012). These genes encode viral transcription factors (BZLF1), a viral DNA polymerase (BALF5) and associated factors, viral glycoproteins (gp350/220 and gp110), and structural proteins (capsid and tegument proteins) (Kanda et al., 2019). Furthermore, the proteins involved in the lytic cycle are categorized into three groups according to the stage in which they are involved. BZLF1 and BRLF1 are the two proteins that take part in immediate-early replication. The early replication stage encompasses BMRF1, BALF5, BGLF4, and BHRF1. BCRF1, BNRF1, and BFRF3 are involved in the late replication phase.
Against this conceptual background, we note that the host-virus Protein–Protein Interactions (PPIs) play pivotal roles in Biological Processes (BP) crucial for viral pathogenesis and by extension, inform antiviral drug discovery and therapeutics innovations. Viruses depend on host proteins to enter host cells for replication. This leads to alterations in normal signaling mechanisms, resulting in pathological conditions.
In the case of the EBV–human PPI map, most of the interactome is diversely spread and not assembled adequately. Such a comprehensive PPI network of EBV-human can reveal more about the host mechanisms that the virus exploits at different stages. In this study, we report a comprehensive map of the EBV–human protein interactions, encompassing 1752 human and 61 EBV proteins. We assembled the EBV–human PPI data from databases and published proteome data of B cells induced by EBV. This allowed for the most comprehensive PPI map of EBV-human protein to be developed, identifying highly connected proteins in the interaction network. Furthermore, by utilizing drug–protein interaction data, FDA-approved drugs that can potentially target those proteins are identified. Together, this knowledge can be utilized to predict and distinguish significant regulators of both cycles, which could serve as future drug targets contributing to the development of combinatorial therapies against EBV infection.
Materials and Methods
Retrieval of EBV–human PPI data
EBV-human PPI were obtained from HPIDB (v3.0), a significant consortium dedicated to host–pathogen interactions (Hingley et al., 1986). This dataset provides comprehensive information, including identifiers, detection methods, interaction types, confidence values, and corresponding sequences for both human and EBV proteins. We extracted multilevel host–pathogen PPIs for subsequent analysis. As a preliminary measure, we standardized the protein IDs using the UniProt batch query, converting them into their respective original gene symbols for both humans and EBV to ensure uniformity and prevent the loss of any relevant data (UniProt, 2021).
Proteomics data curation
The curation of research articles was carried out to identify proteins with differential expression in humans infected by EBV. We conducted a PubMed search using specific keywords, including “Epstein–Barr Virus” AND “proteomics” AND “Homo Sapiens” AND “blood” NOT “Review.” Out of the initial 72 articles, we focused on those exclusively employing mass spectrometry, resulting in a selection of ∼9 research articles for further investigation (Table 1). In our study, we specifically analyzed experiments conducted solely in B-cells.
Curated Research Articles
Gene ontology and pathway enrichment analysis
The functional enrichment analysis of human proteins identified from HPIDB (v3.0) and those that were differentially expressed due to EBV infection was conducted using the ShinyGO tool (http://ge-lab.org/go/) (Ge et al., 2020). This analysis involved exploring BP, Molecular Function (MF), Cellular Components (CC), and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways associated with the respective genes. False Discovery Rate (FDR) is a statistical method for multiple-testing correction that considers the number of tests and an uncorrected p-value threshold to evaluate the fraction of falsely enriched pathways over enriched pathways (Farooq et al., 2020). FDR <0.05 was applied to determine statistically significant enrichment results for the analysis.
EBV–human PPI network construction
All the curated differentially expressed proteins, encompassing both upregulated and downregulated ones, were further considered for evaluating PPIs after excluding duplicate values using the String database (https://string-db.org/) (Szklarczyk et al., 2023). Further, we deselected “text mining” in the active interaction source tab, hid the “disconnected nodes in the network” in the “network display option,” and set the confidence value to “high” (0.70). The human–EBV PPI data retrieved from HPIDB was integrated into Cytoscape (version 3.10.0) (Shannon et al., 2003) initially to generate a primary PPI network. Furthermore, the PPI data provided by STRING were imported into Cytoscape to integrate it with the primary network, developing a comprehensive virus–human PPI network.
Identification of highly connected proteins from the network
The CytoHubba plugin was utilized to identify highly connected proteins within the network. CytoHubba provides 11 different topological analysis methods for interpreting important nodes in a biological network (Chin et al., 2014). In this study, the degree parameter was employed to analyze the network, with a higher degree value indicating a greater number of interactions with other proteins. Basically, this method is locally based and considers only the direct neighborhood of a vertex. Given a node v, N (v) denotes the collection of its neighbors. The degree method (Deg) is defined as Deg (v) = |N (v)|. The top 20 human proteins, ranked as highly connected proteins, were selected for further analysis.
Cluster analysis
Cluster analysis was conducted to detect and group highly interconnected proteins with similar functions or those related to similar pathways. The Molecular Complex Detection (MCODE) plugin was utilized to identify gene clusters within the network. This method assigns weights to vertices based on the density of their immediate surroundings (Bader and Hogue, 2003). The parameters were set to default values, including a Degree Cutoff of 2, Node Score of 0.2, K-Core of 2, and Max.Depth of 100.
Identification of intrinsically disordered proteins
Proteins that directly interact with EBV proteins were analyzed for structural-related disorders using the DisProt platform, a database of manually curated Intrinsically Disordered Proteins (IDPs) (https://disprot.org/) (Hatos et al., 2020). IDPs specific to the human organism were identified which are involved in the primary interaction network. Cancer-related disordered proteins were then retrieved and searched against the identified disordered proteins data to extract cancer-specific disordered proteins that interacted with EBV proteins.
Construction of drug–protein interaction network
The drug–protein interaction network was constructed utilizing the Drug Gene Interactions Database (DGIdb) (https://www.dgidb.org). This online database encompasses information on all drug–gene interactions from various databases, publications, and other web-based resources related to the gene lists we uploaded (Wagner et al., 2016).
Results
Construction of comprehensive EBV–human PPI network
To visualize the molecular interaction network influenced by EBV infection in humans, we initially retrieved EBV–human PPIs from the HPIDB (v3.0) database and imported them into Cytoscape. This dataset comprises 2276 unique interactions involving 61 EBV proteins and 1058 human proteins. Analysis of manually curated differentially expressed proteins specific to human B cell-mediated EBV infection revealed 1286 unique proteins, of which 766 were upregulated and 520 showed downregulation. To elucidate the interactions among these differentially regulated proteins, we utilized the STRING database and constructed a network. The primary EBV–human PPI network contained interaction data from HPIDB. The differentially expressed proteins interaction network generated by STRING was then imported into Cytoscape to develop the comprehensive EBV–human interaction network, consisting of 1813 nodes and 8657 edges (Fig. 1) (Supplementary Table S1).

Comprehensive EBV–human PPI network generated using Cytoscape. The network includes 1813 proteins and 8657 interactions, consisting of 61 EBV proteins and 1752 human proteins. EBV latent proteins, lytic proteins, primary human protein interactors, and secondary interactors from String database are displayed in varying shades of grey. EBV, Epstein Barr virus; PPI, Protein–Protein Interaction.
Considering that EBV infection in humans involves two main stages, latent and lytic, we further categorized the comprehensive PPI network based on the presence of EBV proteins in these stages. This categorization facilitates an in-depth proteomic analysis of EBV proteins involved in both stages and their interacting partners. In the comprehensive PPI network, out of 61 EBV proteins, 9 are involved in the lytic stage of infection (BCRF1, BDLF3, BGLF4, BHRF1, BLLF1, BLRF2, BNRF1, BRLF1, and BZLF1) and 6 are associated with the latent stage of infection (EBNA1, EBNA2, EBNA3, EBNA-LP, LMP1, and LMP2) (Nagata and Hayashi, 2020). This results in two independent interaction networks with primary and secondary interactor host proteins. These networks were also constructed in Cytoscape, comprising 753 nodes and 3260 edges in the latent stage network (Fig. 2A) (Supplementary Table S2a), while the lytic network is composed of 490 edges and 612 nodes (Fig. 2B) (Supplementary Table S2b). This indicates that latent infection exerts a greater influence on human pathogenesis through human–protein interactions.

Latent and lytic EBV–human PPI networks generated using Cytoscape.
Functional enrichment analysis
To explore the functional role of human proteins directly influenced by EBV proteins in lytic and latent stages of infection, we conducted Gene Ontology (GO) and KEGG pathway enrichment analysis. The construction of the PPI network identified 747 and 481 human proteins present in latent and lytic stages, respectively, which were further subjected to GO analysis using the ShinyGO tool. The results highlight the top enriched proteins corresponding to BP, MFs, and KEGG pathways.
The gene enrichment analysis of the human proteins involved in the latent EBV infection stage indicated that viral processes, mRNA metabolic and catabolic processes, and cellular macromolecular catabolic processes are highly enriched BP during the infection. Their MFs include RNA binding, structural constituent of ribosome, protein-containing complex binding, structural molecule activity, and nucleoside-triphosphatase activity. The KEGG pathway enrichment analysis results indicated that ribosome, Coronavirus disease, proteasome, Parkinson's disease, and EBV infection pathways were highly enriched (Fig. 3A).

Bubble plots indicating GO and KEGG pathway enrichment analysis of
Enrichment analysis of human proteins involved in lytic PPI network revealed their significant involvement in BP such as cytoplasmic translation, viral processes, establishment of protein localization to organelle, cellular protein localization, and cellular macromolecular localization. MF enrichment results indicated associations with RNA binding, cell adhesion molecule binding, structural constituent of ribosome, structural molecule activity, and protein-containing complex binding. Pathway enrichment analysis using KEGG identified various biological pathways, including ribosome, Coronavirus disease, Amyotrophic lateral sclerosis, and pathways of neurodegeneration (Fig. 3B).
Identification of highly connected proteins
The CytoHubba plug-in within Cytoscape was employed to identify highly connected hub proteins in both latent and lytic EBV–human PPI networks. The top 20 proteins, exhibiting the highest degree of connectivity, were subsequently retrieved from the networks for further analysis. All 20 highly connected proteins in the latent PPI network showed a network score greater than 90. Among them, RPS3A (Small Ribosomal Subunit Protein eS1) ranked as the top-connected protein with a node score of 120. The result further indicated that most of the proteins within the first 20 are mainly involved in Ribosomal Protein (RP) L (Large) and S (Small) classes. In contrast, the top 20 highly connected proteins present in lytic PPI networks have a network score ranging from 90 to 6. RACK1 (receptor for activated C kinase 1) scored highest with a score of 90 within them. Most of the highly connected proteins in the lytic PPI network were identified in the category of actin alpha, beta, and gamma, chaperones containing subunit, cyclin-dependent kinase, and fibronectin proteins (Table 2).
Human Highly Interacted Proteins Identified in Latent and Lytic Stages of Epstein–Barr Virus Infection
EBV–human PPI network-based cluster analysis
Clustering analysis was performed for isolating interacting proteins that participate in the same biological functions and specific biological pathways. The proteins involved in latent and lytic EBV infection stages were independently analyzed using the MCODE plug-in to generate clusters. In the latent network, 11 clusters were identified, of which the top 3 clusters (Fig. 4A–C), with a cluster score ranging from 35 to 5, were considered for further analysis. Conversely, only one cluster (Fig. 4D) was identified in the lytic network with a score around 3.9. KEGG pathway enrichment analysis of the top 3 clusters in the latent network indicates that proteins in Cluster 1 are highly enriched in ribosome and Coronavirus disease. Cluster 2 proteins show high enrichment in pathways associated with proteasome, EBV infection, Parkinson's disease, Prion disease, Huntington's disease, Amyotrophic lateral sclerosis, Alzheimer's disease, and pathways of neurodegeneration. Meanwhile, Cluster 3 showed only one enrichment with spliceosome-associated pathways (Fig. 5A). In the case of the lytic network, pathway enrichment results revealed that they are highly enriched in apoptosis, Prion disease, protein processing in the endoplasmic reticulum, lipid and atherosclerosis, Parkinson's disease, and Amyotrophic lateral sclerosis-associated pathways (Fig. 5B).
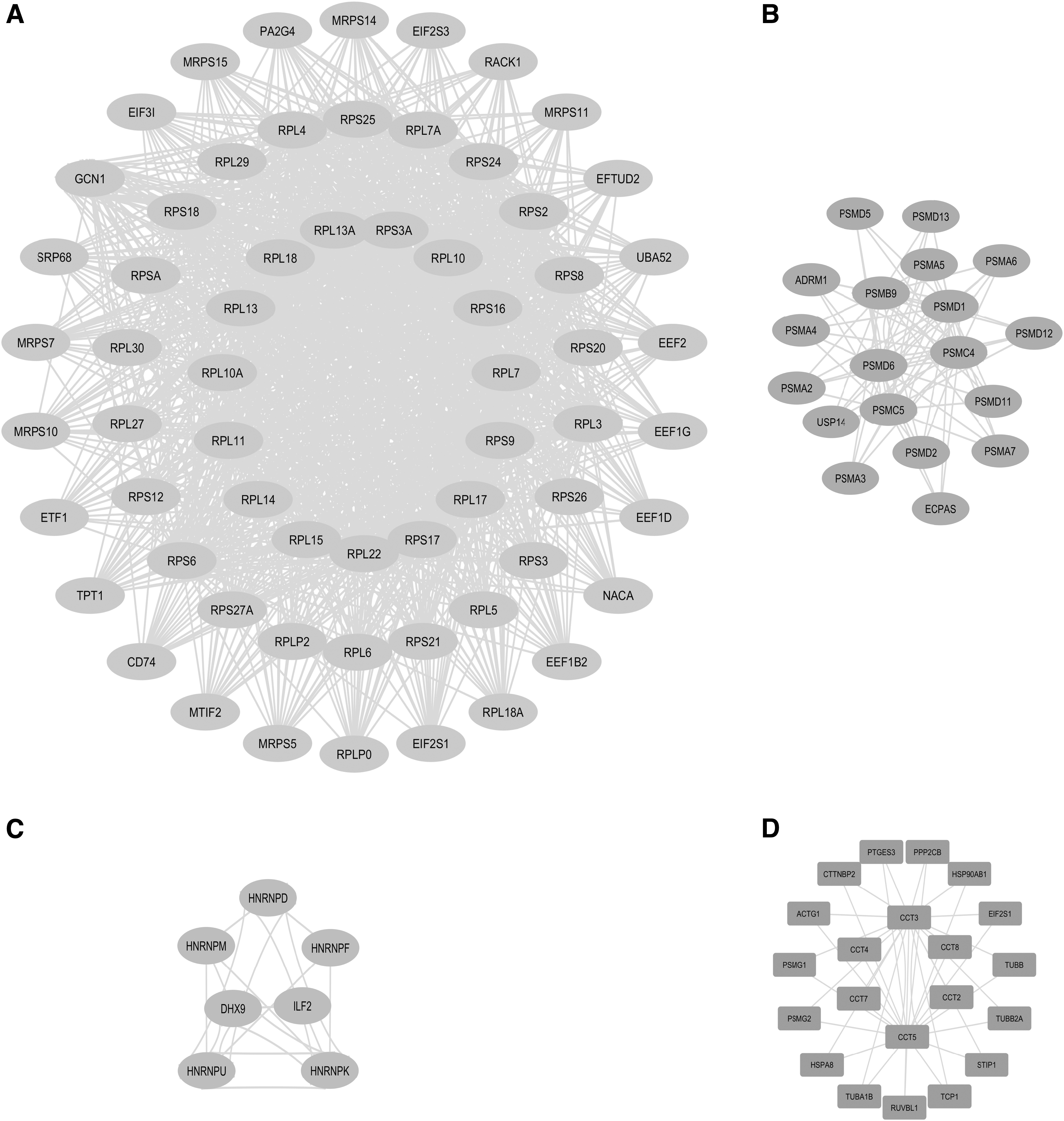
Clusters identified in latent and lytic PPI networks using MCODE plug-in. Top 3 clusters in latent network including

Bubble plot showing KEGG pathway enrichment analysis of clusters using ShinyGO.
Identification of intrinsically disordered and cancer-specific proteins
IDPs in humans play a major role in virus pathogenicity and infectivity, implicating numerous biological functions. The analysis of IDPs revealed that both EBV latent and lytic proteins contain a substantial number of disordered proteins, with ∼20% being specific to cancer. The study identified 119 IDPs within the primary interactors of EBV proteins, and among them, 24 belong to cancer-related disordered proteins (Supplementary Table S3a). In the latent PPI network, 66 proteins were identified as intrinsically disordered, and among them, 16 proteins (GMPS, MYC, SND1, TGFBR2, NPM1, CDKN1A, SRC, SF3B1, HNRNPA2B1, TP53, HMGA2, MDM2, EP300, RB1, UBR5, and LCK) showed specificity to cancer (Supplementary Table S3b). In contrast, the lytic PPI network involved 38 IDPs, and among them, 11 (NPM1, DAXX, XPC, UBR5, MYC, CREBBP, SRC, EP300, TP53, BAX, and EGFR) are more specific to cancer (Supplementary Table S3c). It was identified that six IDPs associated with cancer (MYC, NPM1, SRC, TP53, EP300 and UBR5) were found to be present in both latent and lytic PPI networks (Supplementary Table S3d).
Drug–protein interaction analysis
Human proteins with an interaction cut-off score of 20 among the highly connected proteins identified from the lytic and latent PPI network underwent drug–protein interaction analysis using the DGIdb tool. The findings indicated that within the latent network, only one protein, namely RPL13, displayed interactions with FDA-approved drugs. These drugs include Thalidomide and Docetaxel, both of which are utilized in the treatment of cancer. Similarly, among the highly interacted proteins in the lytic PPI network, two of them (PSMC4 and ITGB1) demonstrated interactions with drugs. ITGB1 showed a higher number of drug interactions with anticancer drugs such as Volociximab, Etaracizumab, Abituzumab, and some other drugs used for the treatment against multiple sclerosis, including Firategrast and Natalizumab. On the other hand, the PSMC4 protein exhibited interactions with four anticancer drugs, specifically Carfilzomib, Bortezomib, Ixazomib Citrate, and Oprozomib (Table 3).
Highly Connected Human Proteins Showing Interaction with FDA-Approved Drugs Obtained from Drug–Gene Interactions Database
EBV, Epstein Barr Virus.
Discussion
The global threat posed by EBV infection, claiming ∼160,000 lives each year (Khan et al., 2020), has become a significant concern. The persistent latent infection, high mutation rate, and the absence of effective vaccines have collectively rendered EBV a longstanding epidemic disaster (Odumade et al., 2011). Our study aims to understand the roles of viral proteins interacting with human host proteins, and their in depth analysis on different infection stages. Investigating these interactions, both individually and in the context of human protein interactomes, provides crucial insights into EBV pathogenesis. Despite available virus-host PPI networks for various viruses, there's a lack of an extensive EBV-human PPI network (Calderwood et al., 2007).
To address this gap, the first step involves collecting EBV-interacted human protein data from diverse databases and assembling EBV-infected B cells' specific proteomic information. Second, recognizing two distinct infection stages, lytic and latent, it is crucial to represent their specific molecular associations separately. Recognizing the need for a comprehensive EBV-human PPI to understand altered interaction networks, this study integrates dysregulated proteomic information from published literature and specific database entries. Interestingly, the PPI network established through the integration of HPIDB and curated data yielded a total of 8657 interactions, encompassing 61 EBV proteins and 1752 human proteins. Additionally, the categorization of this entire network into latent and lytic stages identified the presence of 753 proteins and 3260 in the latent and 490 proteins and 612 interactions in the lytic PPI networks. This promises to enhance proteomic analysis at different stages of EBV infection and pathogenesis.
The functional enrichment analysis of human proteins associated with latent and lytic stages of EBV infection provides in-depth information about the signaling pathways and cellular processes intercepted by EBV infection. It is identified that most proteins participating in latent and lytic PPI networks are associated with BP such as mRNA catabolic processes, viral processes, and cytoplasmic translation. One of the reasons for this enrichment is the degradation of host mRNAs, competing with viral mRNAs for translation and ensuring the efficient translation of viral transcripts (Stern-Ginossar et al., 2019; Walsh and Mohr, 2011). Furthermore, EBV manipulates cellular macromolecule catabolic processes, including protein degradation, to favor the production of viral proteins and virions. This manipulation enables the virus to evade host immune responses and facilitate viral replication (Kalla et al., 2012; Rosemarie and Sugden, 2020). Additionally, studies have reported that human eukaryotic initiation factors, regulated by viral proteins, play a role in the translation initiation of cytoplasmic components, indicating viral control over host gene replication (Liu et al., 2020; Lloyd, 2006).
MF enrichment highlights the significant involvement of proteins in two stages in RNA binding, structural constituent of ribosome, protein-containing complex binding, structural molecule activity, and nucleoside-triphosphatase activity. This is attributed to the utilization of host CC by EBV to interact with RPs and translation factors, promoting the synthesis of viral proteins (Alenquer and Amorim, 2015; Walsh and Mohr, 2011). Some of the EBV-induced human proteins in both the networks, namely chaperonin containing TCP1 subunits (CCT2, CCT3, CCT4, CCT5, CCT6A, CCT7 and CCT8), heat shock proteins (HSP90AA1, HSP90AB1, HSP90B1, HSPA5, HSPA8 and HSPD1), proteasome subunits (PSMC1, PSMC2, PSMC3, PSMC5, PSMC6, and PSMD6), and tubulin proteins (TUBA1A, TUBA1B, TUBA4A, TUBB1, TUBB2A, TUBB4A, TUBB4B and TUBB6) possess nucleoside-triphosphatase activity, involved in processes like DNA replication and nucleotide metabolism. These activities contribute to the virus's ability to replicate and maintain its genome within host cells (Meng et al., 2010).
KEGG pathway enrichment analysis identifies that latent and lytic-induced human proteins that are highly associated with virus disease, Amyotrophic lateral sclerosis, ribosomes, neurodegeneration, Parkinson's disease, Huntington's disease, and viral carcinogenesis-related pathways. Proteasome subunits, Calcium Dependent Kinase (CDK), histone clusters, Tumor Necrosis Factor (TNF) receptor-associated factors, tyrosine activation proteins, and RPs are implicated in these pathways. Studies have shown that EBV infection can directly or indirectly infect neurons via infected B-lymphocytes, inducing neuroinflammation and demyelination. This promotes the proliferation, degeneration, and necrosis of glial cells, further leading to proliferative disorders of B- and T-lymphocytes, and the development of nervous system diseases, such as Alzheimer's disease, Parkinson's disease, multiple sclerosis, acute cerebellar ataxia, meningitis, acute disseminated encephalomyelitis, and brain tumors (Houen et al., 2020; Huang et al., 2021; Leta et al., 2022; Zhang et al., 2021). Additionally, Shannon-Lowe et al. (2017) explored the role of EBV in BL, a type of non-Hodgkin lymphoma that can manifest as acute leukemia. Characterized by the overgrowth of B cells, this condition is often associated with EBV infection, especially in regions where the virus is prevalent (Brady et al., 2007; Lopez et al., 2022). EBV is known to infect B lymphocytes and can contribute to the development of malignancies by altering the normal regulation of cell growth (Krishna et al., 2023; Krishna et al., 2020; Saha and Robertson, 2019; Sugimoto et al., 2023). In the context of leukemia, EBV has been found to play a role in the transformation of B cells, leading to uncontrolled cell proliferation (Price et al., 2017).
Furthermore, cluster analysis of the PPI networks identified interacting proteins in both networks that are involved in the same biological pathways. In the latent PPI network, the cluster subunits are associated with ribosomal, proteosomal, and ribonucleoprotein pathways. Studies have reported that during EBV infection, RPs like RACK1 interact with different signaling proteins and activate various signaling events (Miller et al., 2021). The proteasome plays a role in modulating the degradation of viral proteins, influencing the progression and outcomes of EBV infection, and has been implicated in various neurodegenerative diseases, including Parkinson's disease, Prion disease, and Huntington's disease (Kaleli et al., 2020). In the lytic PPI network, clusters are enriched with multiple pathways, and the dysregulation of these interconnected pathways by EBV could contribute to the pathogenesis and progression of various diseases, emphasizing the complexity of virus–host interactions in impacting cellular homeostasis and disease outcomes (Trivedi et al., 2021).
Highly connected proteins in a PPI network confirm their likely pivotal role in the progression and pathogenesis of EBV infection, given their involvement in multiple pathways. Notably, all the top 20 highly connected proteins in the latent PPI network are associated with ribosomes, their biogenesis, RNA binding, and cytoplasmic translation. Studies indicate the critical role of continued RP synthesis in maintaining viral persistence and latency during HSV-1 infection, a herpesvirus related to EBV (Greco et al., 1997; Simonin et al., 1997). This further suggests that the RPs identified in our study may contribute to promoting viral mRNA translation by ensuring sustained synthesis during latent EBV infection (Sim and Talwar, 2019). On the other hand, hub proteins in the lytic PPI network belong to different categories, including kinase receptors, chaperone-containing subunits, proteasome subunits, integrin subunits, Adenosine Triphosphate (ATPase), polymerases, and oncogenes.
Analysis of IDPs intriguingly identified their presence in both stages of EBV infection. Among the 119 identified IDPs, 38 belong to the lytic PPI network, and 66 belong to the latent PPI network. Hub proteins ITGB1 and P4HB in the lytic PPI network, as well as RPL4 and RPL10 in the latent PPI network, were also found to be intrinsically disordered structures. Furthermore, 19 disordered proteins (ADRM1, CNOT1, DDX6, EMD, EP300, HSPA5, ITGB1, LOX, MYC, NPM1, P4HB, RPA1, RPL10, RPL4, SRC, TP53, TUBA1B, UBR5, and XRCC5) were identified in both networks. The disordered structural conformations of these proteins, as a primary interactor of viral proteins, indicate a higher chance of the development of diseases such as cancer and other malignancies. Moreover, out of the 24 cancer-specific IDPs, 6 of them (UBR5, EP300, TP53, MYC, SRC, and NPM1) were identified in both PPI networks. These proteins are associated with thyroid cancer, endometrial cancer, chronic myeloid leukemia, and Kaposi sarcoma-associated herpesvirus infection (Qiu et al., 2022; Sobocan et al., 2020; Williams et al., 1999). Studies have indicated that two distinct disordered proteins in the latent network, CDKN1A and TGFBR2, exhibited differential expression in patients with breast cancer (Lin et al., 2017; Wei et al., 2015). Additionally, specific disordered proteins from the lytic PPI network also demonstrated oncogenic properties. For instance, the DAXX protein was found to promote tumorigenesis and disease progression in patients with prostate cancer (Mahmud and Liao, 2019). Another well-known protein, EGFR, serves as a driver of tumorigenesis in lung and breast cancer, as well as in glioblastomas (Sigismund et al., 2018).
Through an analysis of drug–protein interactions, potential drug candidates binding to highly connected proteins in two PPI networks were identified. RPL13, a central highly connected protein in the latent PPI network, showed interactions with anticancer drugs such as Thalidomide and Docetaxel. Thalidomide, primarily used for skin diseases associated with leprosy, has also been studied for treating multiple myelomas (Franks et al., 2004; Kim and Scialli, 2011). Docetaxel, an intravenous drug, is used alone or in combination for various cancers (Fulton and Spencer, 1996). In contrast, proteins like PSMC4 and ITGB1 in the lytic PPI network interact with antiviral, anticancer, and anti-sclerosis drugs, as well as antibodies. Natalizumab, a major multiple sclerosis drug, interacts with the ITGB1 protein. Etaracizumab, an anticancer drug, is used for renal cancer treatment and has potential in reducing ovarian cancer proliferation and invasion (Landen et al., 2008). Antibodies like Volociximab, used alone or with chemotherapy, and Abituzumab, used in combination with Firategrast, an integrin antagonist, aim to reduce lymphocyte trafficking into the Central Nervous System (CNS) and decrease multiple sclerosis activity (Almokadem and Belani, 2012). Studies also indicated that PSMC4 interacts with anticancer drugs including Bortezomib, Ixazomib Citrate, Carfilzomib, and Oprozomib, primarily used for multiple myeloma (Cengiz Seval and Beksac, 2018; Offidani et al., 2014; Scott et al., 2016; Shah et al., 2019). Notably, Bortezomib efficiently targets gammaherpesvirus-associated B cell lymphomas triggered by EBV viral lytic cycle activation, suggesting potential therapeutic benefits in patients affected by gammaherpesvirus-associated lymphomas (Granato et al., 2017).
Though the comprehensive EBV–human protein interaction map provides new insight into the pattern of EBV-human PPI during lytic and latent stages, the study does have the following limitations: (1) Even though EBV encodes around 89 proteins, the study accounts 61 EBV proteins and their interactions with human proteins due to the unavailability of comprehensive data in HPIDB and curated proteomic datasets. (2) The curated proteomic data utilized in the study are specific to B cells, the primary target of EBV. This results in the omission of interactions occurring in other cell types during EBV infection. (3) The interaction map does not explore details about the regulatory mechanisms governing the identified interactions, such as posttranslational modifications. (4) While the study emphasizes the impact of EBV proteins on cancer and suggesting potential drug repurposing strategies, additional experimental validations are imperative to confirm these findings further and establish the efficacy of such therapeutic interventions.
Conclusions
This study offers a novel perspective on the intricate PPIs between EBV and the human host. The subdivision of the comprehensive network into lytic and latent PPI networks has revealed distinctive interaction patterns in both stages, shedding light on their respective effects on the host system. The identified clusters within the PPI networks provide detailed insights into the regulation of specific proteins, elucidating their associated functions and pathways relevant to EBV-mediated cancers and other diseases.
Notably, the study highlights the significance of various intrinsically disordered and cancer-related proteins, both commonly and specifically identified during the lytic and latent stages of EBV infection. While identifying highly connected proteins within the networks, including RPL13, ITGB1, and PSMC4, this study underscores their potential interactions with antiviral, anticancer, and antisclerosis drugs. Together, the study unravels detailed PPI between EBV and the human host, providing insights into their regulation of various direct or indirect pathways with stage-level classification. These findings hold timely and notable implications for the prevention and treatment of EBV infections and associated pathologies, offering a foundation for further therapeutics innovation.
Footnotes
Acknowledgments
The authors acknowledge the Center for Systems Biology and Molecular Medicine (CSBMM), Yenepoya Research Centre, Yenepoya (Deemed to be University) for providing the necessary facilities required for conducting this study.
Authors' Contributions
C.S.A. conceptualized and designed the study. D.K., S.B., and C.S.A. performed data curation, analysis, and visualization. Manuscript written by D.K. and C.S.A. and edited by R.R., M.V.V., and T.S.K.P. All authors made a significant intellectual contribution, have read and approved to the final version of the manuscript for publication.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.