
Abstract
MicroRNAs (miRNAs) have emerged as a prominent layer of regulation of gene expression. This article offers the salient and current aspects of machine learning (ML) tools and approaches from genome to phenome in miRNA research. First, we underline that the complexity in the analysis of miRNA function ranges from their modes of biogenesis to the target diversity in diverse biological conditions. Therefore, it is imperative to first ascertain the miRNA coding potential of genomes and understand the regulatory mechanisms of their expression. This knowledge enables the efficient classification of miRNA precursors and the identification of their mature forms and respective target genes. Second, and because one miRNA can target multiple mRNAs and vice versa, another challenge is the assessment of the miRNA-mRNA target interaction network. Furthermore, long-noncoding RNA (lncRNA)and circular RNAs (circRNAs) also contribute to this complexity. ML has been used to tackle these challenges at the high-dimensional data level. The present expert review covers more than 100 tools adopting various ML approaches pertaining to, for example, (1) miRNA promoter prediction, (2) precursor classification, (3) mature miRNA prediction, (4) miRNA target prediction, (5) miRNA- lncRNA and miRNA-circRNA interactions, (6) miRNA-mRNA expression profiling, (7) miRNA regulatory module detection, (8) miRNA-disease association, and (9) miRNA essentiality prediction. Taken together, we unpack, critically examine, and highlight the cutting-edge synergy of ML approaches and miRNA research so as to develop a dynamic and microlevel understanding of human health and diseases.
Introduction
In the current post-genomic and multi-omics era, there is growing emphasis on regulation of gene expression. MicroRNAs (miRNAs) are small endogenous noncoding RNAs with a directive role in the posttranscriptional regulation of gene expression across organisms (Ambros et al., 2003; Bartel, 2004; Lagos-Quintana et al., 2001; Lau et al., 2001). The miRNAs are one of the prevalent groups of gene regulatory systems, impacting the expression of protein-coding genes (Lewis et al., 2003).
The biogenesis of miRNA is tightly regulated, and the dysregulation of this process has been linked to both health and susceptibility to various human diseases, including cancers and infections (Peng and Croce, 2016). This article offers the salient and current aspects of machine learning (ML) tools and approaches from genome to phenome in miRNA research.
Overview of miRNA Integrative Biology
The miRNAs are mostly transcribed from intergenic regions and in a promoter-dependent or -independent manner from intronic regions by RNA polymerase II. The biogenesis of miRNAs is not restricted to a canonical pathway; rather, it has also been proposed through multiple noncanonical pathways (Chong et al., 2010).
In the canonical pathway, the transcribed primary miRNA is processed into a 70-nucleotide hairpin loop structure precursor by ribonuclease III (Drosha) (Lee et al., 2003), a component of two multiprotein complexes. The larger complex comprises a diverse array of RNA-associated proteins, including RNA helicases; the smaller complex contains Drosha and the double-stranded heme-binding protein DGCR8 (DiGeorge Syndrome Critical Region 8) (Faller et al., 2007), which is located in the nucleus and its gene deletion characterizes the DiGeorge syndrome (Gregory et al., 2004). Subsequently, the precursor miRNAs are exported from the nucleus to the cytoplasm mostly by the nuclear export receptor XPO5 (Exportin 5) (Yi et al., 2003).
In the cytoplasm, the precursor miRNA undergoes further processing by a second RNAse III-type endonuclease, Dicer, which cleaves the loop structure to liberate an miRNA duplex with a length of 19–22 nucleotides (Cullen, 2004). In the process of miRNA silencing, the methylated mature miRNA duplex separates, and one strand is selectively bound into a protein complex known as the miRNA-induced silencing complex (miRISC), which consists of multiple subunits, including Argonaute (Lagos-Quintana et al., 2001). The other strand is typically degraded. The miRISC engages the complete or partial complementarity binding of miRNA seed region to the 3′-untranslated region (UTR) of the target mRNA, orchestrates mRNA deadenylation, and decapping toward degradation or translational repression (Krol et al., 2010; Siomi and Siomi, 2010). Noncanonical miRNA biogenesis pathways depend on the source of the primary miRNAs, such as miRNAs encoded in the introns (mirtrons), endogenous short-hairpin RNAs, and tRNA fragments.
Together, they contribute to the miRNA pool, as reviewed elsewhere (Ha and Kim, 2014; Kim, 2005).
MiRNAs have the ability to bind to different regions of mRNA, such as the 5′-UTR and protein-coding exon regions, but the base pairing between the miRNA seed region and the 3′-UTR of mRNA is mainly responsible for the quantitative regulation of mRNAs or their protein forms (Lewis et al., 2005). When miRNAs have highly complementary targets, they can induce mRNA degradation accompanied by tailing and trimming. Interestingly, one miRNA can target multiple mRNAs simultaneously, and the reverse is also true. This results in the formation of complex posttranscriptional gene regulatory networks (Enright et al., 2003).
Emerging research has demonstrated the interaction of miRNAs with their targets, including mRNAs (Liu et al., 2014), long-noncoding RNAs (lncRNAs) (Yoon et al., 2013), circular RNAs (circRNAs) (Hansen et al., 2013), and pseudogenes (Thomson and Dinger, 2016). The lncRNAs can regulate the expression and function of miRNAs by acting as miRNA sponges or competing with miRNAs for binding to their target mRNA. The interaction between miRNA on lncRNA and circRNA can be complex and context-dependent, and understanding the specificity or preferentiality of such interactions can provide insights into the dynamicity of regulatory networks. This introduces the necessity for analyzing the layer of the dynamic regulatory network of the miRNAs and their targets in diverse biological conditions (Paraskevopoulou and Hatzigeorgiou, 2016; Yoon et al., 2014).
The influence of miRNAs extends to a multitude of target genes enabling the regulation of the growth and proliferation of cells, tissue differentiation, and embryonic development. In this regard, it is unsurprising that the imbalance in miRNA-mediated regulation has been associated with the emergence of several types of cancer (Hwang and Mendell, 2006). For example, the differential expression of miR-21 results in ovarian, colorectal, and leukemia cancers (Adams et al., 2014). Identifying and interpreting the functional impact of miRNAs using experimental approaches are challenging, given the complexity of their regulatory target networks in specific biological conditions.
These challenges extend to the analysis of the miRNA coding potential of genomes, prediction of their promoters, characterization of precursor miRNAs, identification of mature miRNA sequences, their potential interactions with noncoding RNAs, analysis of their mRNA target specificity and diversity, and subsequently, the functional characterization of miRNAs in specific biological processes and their association with diseases/disorders (Lauria et al., 2023; Megret et al., 2022). The initial addressing of miRNA for better identification starts from its nomenclature. In miRBase, miRNA nomenclature infers “hsa” as an abbreviation for Homo sapiens, following scientific conventions. This system, based on the initial letters of “Homo” and “sapiens” (h from Homo and sa from sapiens), ensures a standardized and concise representation for human miRNA nomenclature (Griffiths-Jones et al., 2006).
Owing to the limitations in the experimental analysis of miRNA biology increases the essentiality of computational dynamics. Indeed, in the past decade, different computational approaches have been undertaken for unraveling the heterogeneity of miRNAs and their associated molecular pathways. In the past, ML approaches have been used to analyze miRNA turnover, miRNA-target interactions, disease associations, characterize pre- and mature miRNAs, and predict miRNA promoters. Availability of curated experimentally validated miRNAs, their targets, and their association with diseases facilitated by public databases such as miRbase (Kozomara et al., 2019) miRTarBase (Hsu et al., 2011), miRecords (Xiao et al., 2009), Human miRNA disease database (HMDD) (Huang et al., 2019b), and miR2disease (Jiang et al., 2009) has accelerated the progressive development of ML tools.
Most ML-based mRNA target prediction tools, such as TarPmiR (Ding et al., 2016), miRaw (Pla et al., 2018), and miTar (Gu et al., 2021), are designed to use tens to hundreds of different features in their models, such as the sequence, structure, conservation, thermodynamic properties, and context of the interacting molecules. Recently, computationally predicted data architecture has also been accounted for by unsupervised, supervised, and reinforcement learning methods on miRNA biology (Azari et al., 2023; Pawelka et al., 2022).
The availability of new features and data sets of high-throughput information on miRNA-target and other molecular interactions has led to the development of enhanced ML-based methods, such as miES (Song et al., 2019) and miRcorrNet (Yousef et al., 2021). Such tools explore miRNA's involvement in well-known signaling pathways and their associations with diseases. Mining these tools helps to unpack the multilevel prioritization of miRNA. Most tools are made in isolation and must be assembled for effective comparative analysis and visualization by the scientific community to accelerate miRNA-related applications.
The comprehensive information in the following expert analyses offers an extensive compendium of tools and approaches to miRNA, augmented by ML techniques, along with their meticulously stringent comparative evaluation. In structuring our review, we aimed to provide a comprehensive yet focused examination of ML approaches in miRNA research, spanning both the nuclear and cytoplasmic levels of miRNA processing. Our selection of listed items within each topic was guided by the intention to highlight the biological process of miRNA and its interconnected process and how the implementation of these biological factors in an ML perspective. This insight addresses the foundational steps in miRNA biogenesis and regulation, laying the groundwork for subsequent cytoplasmic processing and functional analysis. Thus, our review represents a snapshot of current advancements rather than an exhaustive catalog of all the available approaches.
Genomic and Nuclear-Level Features of miRNA
miRNA prediction in genome
Detecting novel miRNA genes poses a significant challenge in bioinformatics, requiring the integration of various factors such as the characteristic secondary structure of the precursor, the conservation of both primary and precursor sequences, and expression data encoded in small RNA libraries (Hertel et al., 2014).
The search for novel or species-specific miRNAs involves scanning the entire genome or genome-wide alignments with related species using ML approaches. These methods consider various features of the precursor, including its fold-back structure and conservation information, to classify candidate sequences. ML algorithms are trained on sets of known miRNAs especially from the miRBase database (Griffiths-Jones et al., 2006), often incorporating data sets representing sequences with similar characteristics but are not miRNAs. Computational-based tools exist for this purpose, such as miRCheck (Jones-Rhoades, 2010), MapMi (Guerra-Assuncao and Enright, 2010), and miROrtho (Gerlach et al., 2009) differing in their hairpin detection algorithms, feature definitions, and ML approaches. While some tools focus on identifying precursor structures, others prioritize the detection of mature miRNAs by analyzing precursor hairpins and mature miRNA candidates, along with flanking sequence information.
The miRNAs are mostly transcribed by RNA polymerase II and posttranscriptionally modified mainly by 5′capping, splicing, and polyadenylation at the 3′end, to become mature and functional miRNAs (Cai et al., 2004; Lee et al., 2004). For intronic, exonic, or intergenic miRNAs, at the transcription initiation sites, the physical association of polymerase with the promoter are facilitated by transcription factors such as p53 (Tarasov et al., 2007), MYC (Chang et al., 2008b), ZEB1, and ZEB2 (Bracken et al., 2008) and epigenetic regulators (Bueno et al., 2008; Kim et al., 2009). DNA methylation and histone modifications contribute to the regulation of miRNA gene expression (Davis-Dusenbery and Hata, 2010; Lujambio et al., 2008; Scott et al., 2006).
These regulative features, such as histone modification and nucleosome positioning patterns, are used for the computational prediction of miRNA biogenesis (Ozsolak et al., 2008). In this context of identification of polyadenylated miRNA, transcripts have been identified as expressed sequence tags (ESTs) (Chien et al., 2011). Considerable work has been put into developing computational algorithms that integrate chromatin immunoprecipitation (ChIP)-seq data, deoxyribonuclease sequencing, and transcriptome profiles to decode transcriptional-level miRNA regulations (Hollbacher et al., 2020).
The primary miRNA hairpin contains a long imperfect stem of ∼30 bp with a single-/double-stranded junction (Han et al., 2006). Multiple mismatches and wobble base pairs in the upper stem of primary miRNA are considered to analyze the efficiency and accuracy of their processing (Li et al., 2020b). The characterization of precursor miRNA to 60–120 nucleotides by Drosha and DGCR8 accounted for sequence length, number of base pairings, and the free energy for the secondary structure (Starega-Roslan et al., 2011). The unstable nature of the secondary structure depicts that precursor miRNA may be degraded before it can be processed (Suzuki et al., 2011). The presence of stabilizing sequences such as GC regions or hairpin loops and RNA binding proteins necessitates stability (Bail et al., 2010). Figure 1 shows a schematic representation of the miRNA biogenesis in the nuclear level and its associated tools and approaches.
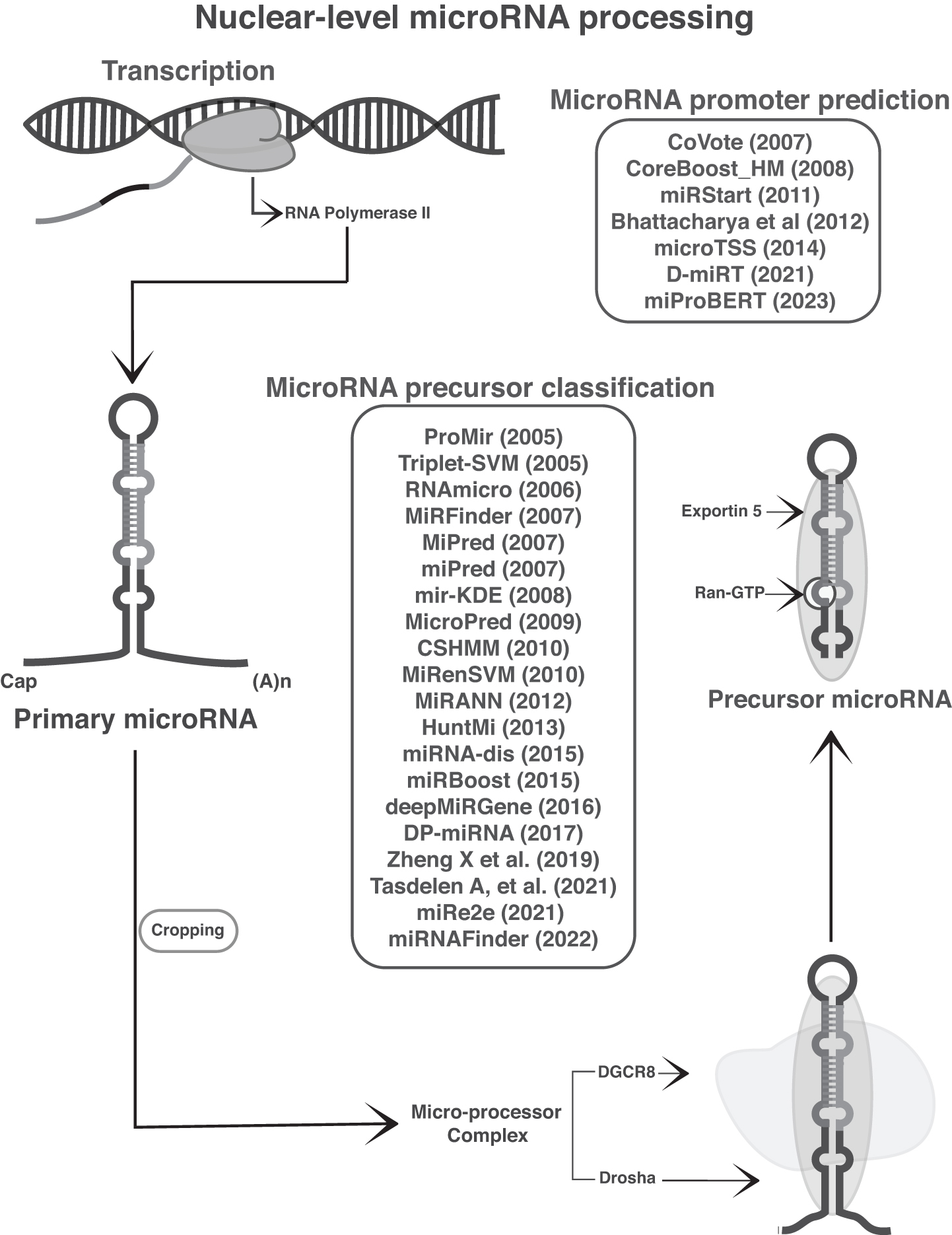
Machine learning-based tools and approaches to processing microRNA biogenesis at the nuclear level. From transcription to precursor microRNA level. Tools/approaches and corresponding processes are also included herein. The year of publication is given in brackets. Some approaches do not have names, so we refer to them with the attendant authors' names.
miRNA promoter prediction
The promoters are essential regions accounted for by different regulatory factors to regulate transcription mostly by RNA polymerase II. Mapping the precise locations of the miRNA promoter regions is crucial for revealing the transcriptional regulatory interface (Weis and Reinberg, 1992; Zeng et al., 2009). Transcription factor-miRNA regulation databases such as TransmiR v2.0 (Tong et al., 2019), microPIR (Piriyapongsa et al., 2012), and miRT (Bhattacharyya et al., 2012a) facilitate the data set collection. The traditional prediction methods have used several genome sequence features, including collective analysis of ESTs and different genome sequence patterns such as TATA box, CAAT box, and GC box (Abeel et al., 2008; Bajic et al., 2004; Ohler and Niemann, 2001).
The culmination of modern technologies results in ML tools; the primary one common query voting (CoVote) used nucleotide sequence motif features in terms of k-mers by building a discriminative model to distinguish polymerase II and polymerase III promoters. Finding the possible k-mers with the help of WordSpy motif-finding algorithm analysis of sequence motifs based on base-pair length results in a support vector machine (SVM)-based classifier (Zhou et al., 2007).
Regarding text data vectorization, the evolution of word embedding methods has considerable untapped potential in addressing sequence-related issues. Similar kinds of traditional nucleotide sequence formats and the addition of CpG island features have been incorporated in another SVM-based approach developed by Bhattacharyya et al. (2012b). Subsequently, CoreBoost_HM, a boosting algorithm approach developed in 2009, used more specific properties regarding gene-regulation epigenetic control markers such as histone markers, acetylation, and methylation signatures and previously adopted CpG island features instead of k-mers (Wang et al., 2009b).
Five years later, the SVM-based miRNA transcription start site prediction tool further integrated high-resolution RNA-sequencing data with active transcription sites derived from ChIP-seq and deoxyribonuclease sequencing, which has revealed dicistronic and polycistronic miRNA transcripts (Georgakilas et al., 2014).
Recently, D-miRT, a neural network-based tool, contributed high-resolution features on the basis of single-base resolution of nucleotides (Cha et al., 2021). In addition, there has been a surprising fact that natural language processing algorithms specifically bidirectional encoder representations from transformers (BERT) lead to a new approach called miProBERT (Wang et al., 2023). The model utilizes the pretrained DNABERT and fine-tunes it on the gene promoter data set to include information about the biological properties of promoter sequences. The model is then used to scan the upstream regions of intergenic miRNAs, resulting in the identification of 665 miRNA promoters. The model's discriminative ability is improved by using a random substitution strategy to construct a negative data set, which reduces the false-positive rate to 0.0421.
On independent data sets, miProBERT outperforms other gene promoter prediction methods and is shown to have a precision of 78.13% and recall of 75.76%. An overview of these algorithms or approaches is summarized in Table 1.
MicroRNA-Promoter Prediction Tools
BERT, bidirectional encoder representations from transformers; ChIP, chromatin immunoprecipitation; CoVote, common query voting; microTSS, miRNA transcription start site.
miRNA precursor classification
Intermediary precursor miRNA and its hairpin loop structures have a major importance in miRNA biogenesis. Computational techniques for miRNA prediction use comparative genomic methodologies such as homology-based searches (Berezikov et al., 2005), genome-wide scanning (Yones et al., 2018), phylogenetic footprinting (Gao et al., 2013), and clustering analysis (Sewer et al., 2005) to find potential precursor miRNAs from candidate hairpins. Classification or prediction of the precursor miRNAs is challenging as many sequences (over 11 million estimated) can fold into precursor miRNA-like hairpin secondary structures (Bentwich et al., 2005). Many of these could be predictably pseudohairpins and require efficient classification. The exponential growth of databases such as miRGen (Megraw et al., 2007) and miRCarta (Backes et al., 2018) has contributed to data sets for the training and testing aspects in ML methods.
The initial methods for classifying the precursor miRNAs account for distant interactions between nucleotides of known genomic sequences. Among these methods, the basic ML hidden Markov model-based tool ProMiR (Sewer et al., 2005) and the context-sensitive hidden Markov model (CSHMM) (Agarwal et al., 2010) use free energy, structural and sequential in terms of the stem's length, number, and proportion of nucleotides. The hidden Markov model is a formal foundation for making probabilistic models of linear sequence labeling problems. During the initial time HMM serves for a diverse range of problems, including genefinding, profile searches, multiple sequence alignment, and regulatory site identification. Most classification models are built by the supervised learning model SVM, which includes Triplet-SVM (Xue et al., 2005), RNAmicro (Hertel and Stadler, 2006), miPred (Jiang et al., 2007), microPred (Batuwita and Palade, 2009), MiRenSVM (Ding et al., 2010), miRNA-dis (Liu et al., 2015), and miRBoost (Tran Vdu et al., 2015).
The mentioned classification models are all utilized by finding the hyperplane that best separated the input data into different classes. The deployment of these tools is based on the information of structured sequence by analyzing the continuously paired or unpaired status of the nucleotide of precursor miRNA and pseudo-pre-miRNA.
The continuously paired nucleotides are part of a stem structure consisting of base pairs that are complementary to each other and form a double-stranded region of the precursor miRNA molecule. In contrast, unpaired nucleotides are part of a loop structure that forms a single-stranded region of the precursor miRNA molecule. The accuracy of these models varies, but they have generally shown 85% above in prediction tasks. Incorporating similar features such as the minimum free energy, local contiguous triplet structure composition, and dinucleotide shuffling results in random forest methods MiPred (Jiang et al., 2007) and HuntMi (Gudys et al., 2013).
Random forest works by combining a multitude of decision trees at training time and outputting the class by the mean prediction of the individual trees. The MiPred can classify precursor miRNA from other noncoding RNAs and protein-coding mRNAs with an accuracy of 92.5 on a set of experimentally validated targets, while HuntMi is designed to identify precursor miRNA from genome sequences by incorporating additional features such as sequence motifs and evolutionary conservation parameters.
Subsequent advancement in the neural networks further contributes many tools in the field of miRNA precursor classification resulting in the development of various tools such as MiRANN (Rahman et al., 2012), deepMiRGene (Seunghyun et al., 2016), DP-miRNA (Tasdelen and Sen, 2021; Thomas et al., 2017; Zheng et al., 2019b), miRNAFinder (Lokuge et al., 2022), and miRe2e (Raad et al., 2022). The accuracy of these tools can vary depending on the data set and task they were evaluated on. Sequential characteristics of the MiRANN enclose the frequency of dinucleotide pairs, the percentage and the propensity of nucleotide bases, and the structure encompassing the number and length of stem-loops and hairpins.
The deepMiRGene approach learns significant characteristics from data without manual feature engineering and produces a model that can appropriately depict the structural properties of precursor miRNAs with the help of long short-term memory (LSTM) networks. The innovative classification methodology put forth by Zheng et al. (2019b) and Tasdelen and Sen (2021) is predicated upon the combined architecture of convolutional and neural networks. As a corollary, they have utilized the one-hot encoding as a means of text data vectorization. Overall, these tools effectively represent a paradigm shift to identify the structural and thermodynamic features of precursor miRNAs, contributing to the ongoing research in this field. An overview of these algorithms or approaches is summarized in Table 2.
MicroRNA Precursor Classification Tools
CNN, convolutional neural network; CSHMM, context-sensitive hidden Markov model; LSTM, long short-term memory; miRNA, microRNA; RNN, recurrent neural network.
Cytoplasmic-Level Features of miRNA
The nuclear export of precursor miRNA is facilitated by karyopherin exportin 5 in the presence of the Ran-GTP cofactor (Yi et al., 2003). The inhibition of exportin 5 is responsible for downregulated expression and increased nuclear localization of the ribonuclease III superfamily of bidentate nucleases, the dicer, which is required to generate an miRNA duplex (Zeng and Cullen, 2004). Subsequent selective loading of AGO protein results in an effector complex called RNA-induced silencing complex (Okamura et al., 2004). Following miRNA duplex loading, the RISC containing Argonaute (Lagos-Quintana et al., 2001) proteins undergoes the crucial process of passenger strand removal. This intricate mechanism is facilitated by the combined action of endonuclease C3PO and the multimeric complex of translin and its associated protein X (Liu et al., 2009).
Unwinding of the miRNA duplex is a process resulting because of the mismatches at nucleotide positions 2–8 and 12–15 in the guide strand (Kawamata et al., 2009). miRNAs bind to AGO with their 5′end embedded inside the AGO MID domain and the 3′end docked at the AGO PAZ domain (Ma et al., 2005; Schirle and MacRae, 2012). Through partial base pairing, miRNA guides the RISC to target mRNAs (Yang et al., 2020a). The mature miRNA identification by ML parameters accounts for stem-loop structure, conserved terminal nucleotides, seed region, mismatches, and thermodynamic stability (Rojas et al., 2020; Wang et al., 2005). The lower stem-loop contains the cleavage site for the dicer enzyme, a key feature for localizing the mature miRNA (Han et al., 2006; Sacar et al., 2013).
Highly stable miRNA duplexes may be less accessible to the RNA-induced silencing complex and, therefore, less effective in regulating gene expression (Hibio et al., 2012). MiRNA biogenesis in the cytoplasmic level and the associated tools and approaches are represented in Figure 2.

Machine learning-based tools and approaches on microRNA biogenesis at the cytoplasmic level processing. From precursor microRNA to various interactions of microRNA with mRNA, lncRNA, and circRNA. MicroRNA disease association prediction tools and approaches are also included herein. circRNA, circular RNA; lncRNA, long noncoding RNA.
Mature miRNA prediction
Further exploration of miRNA envisages the functional characteristics of mature miRNA. Sequential and structural assessment of mature miRNA using ML revolves around miRNA duplex aspects in terms of base pair counts and bulges on the duplex, and frequency of nucleotides. Because there can be deviations from perfect base pairing in the duplex, resulting in bulges or mismatches. Bulges occur when one strand has an unpaired nucleotide, creating a loop in the duplex. Mismatches occur when two nucleotides on opposing strands do not form a perfect Watson crick base pair. These popular conceptions result in the Bayesian framework, SVM, and random forest-based approaches, listed in Table 3.
Mature MicroRNA Prediction Tools
For example, in the naive Bayes approach MatureBayes, the mature miRNA is represented as a two-dimensional character array containing information about the base composition and structure regarding match and mismatch (Gkirtzou et al., 2010), while in MiRduplexSVM, the nucleotide bases are represented by four binary variables that has an accuracy of 85.4% (Karathanasis et al., 2015). MatureBayes achieved an accuracy of 94.5 on a benchmark data set of human miRNAs, which was comparable with the best performing miRNA prediction tools at the time. The statistical ML approach Bayesian framework allows to quantify uncertainty in the estimates and predictions by using the probability distributions instead of point estimates. Another tool, MatPred, identified mature miRNA within novel pre-miRNAs by considering the duplex window region, lower stem-loop, and minimum free energy. Around 94 features have been extracted from the mature miRNA loci and flanking regions.
The decision function of the MatPred is the SVM with the radial basis function kernel (Li et al., 2015). On the contrary, MiRmat has features such as the size and length of internal loops and the number of bulges in mature sequence regions for dicer site prediction (He et al., 2012). The Maturepred includes position-specific features, stability-related features, minimum free energy, and local contiguous triplet structure (Xuan et al., 2011).
miRNA target prediction
Comprehending the overarching picture of miRNA is sustained through rigorous investigations of the most extensively studied partial complementary interaction with mRNA (Parveen et al., 2019; Piletic and Kunej, 2018). The miRNA recognizes its targets by incomplete base pairing to sequence motifs most often present in the 3′-UTR of their target mRNAs (Bartel, 2009). This partial complementarity inflates the count and quality of potential targets and the development of ML tools (Chen et al., 2019a; Reyes-Herrera and Ficarra, 2012). Computational approaches commence with sequence complementarity regarding seed region (Krek et al., 2005) and thermodynamic potentials in terms of stability of miRNA binding to the target mRNA, which takes the measure as Gibbs free energy (Mathews et al., 1999) and evolutionary conservation such as the keeping of sequence across species (Peterson et al., 2014).
The progressive evolution of manually curated database of experimentally validated miRNA events such as miRbase (Kozomara et al., 2019), MiREDiBase (Marceca et al., 2021), and miRDB (Chen and Wang, 2020) currently explodes the target interaction prediction. The classical RNA secondary structure prediction algorithm extends to dynamic programming-based RNA hybrid that uses minimum free energy hybridization as a parameter for target prediction (Rehmsmeier et al., 2004). Instead of only looking at the free energy score and binding in the 5′score, boosted genetic programming-based TargetBoost has discovered a pattern in the miRNA mRNA binding characteristics (Saetrom et al., 2005). The limitations in the reliability of these approaches for feature selection and data coverage led to the implementation of different models such as SVM, naive Bayes, neural networks, and random forest-based ML approaches, listed in Table 4, because of their analytical capability of nonlinear connections.
MicroRNA Target Prediction Tools
SOM, self-organizing maps.
Considerable accurate output accounts position based, structural, and thermodynamic, and it incorporates single-point mutation, seed pairing, nonseed region secondary structure, conservation, free energy, site accessibility, target site abundance, local AU content, GU wobble in the seed match, and seed pairing stability (Lee and Shin, 2012). The first two to eight nucleotides starting at the 5′end and counting toward the 3′end constitute the seed sequence; either side of the seed sequence refers to the flank sequence (Chipman and Pasquinelli, 2019). For example, the seed properties in terms of the number of bulges, symmetric and asymmetric loops are illustrated as feature sets in the naive Bayes-based NbmiRTar tool that does not rely on conserved sequence features (Yousef et al., 2007).
Data sets having reliable features contribute precision to the output, the SVM-based Targetminer uses a multistage filtering strategy that extracts 90 targeted site context attributes (Bandyopadhyay and Mitra, 2009). SVM is a supervised ML algorithm that works by creating a hyperplane that separates data into different classes based on their features (Noble, 2006). In this context of miRNA target prediction, SVM is trained on a set of known miRNA-target interactions to identify new correlations. One advantage of SVM is that it can also be trained with different kernel functions, such as linear, polynomial, and radial basis functions, to capture nonlinear relationships between miRNA and mRNA sequences. For example, tools such as miTarget (Kim et al., 2006), MirTarget (Wang and El Naqa, 2008), MirTif (Yang et al., 2008), and mirMark (Menor et al., 2014) functioned on this.
The advancements have enabled researchers to design and develop more complex models with deeper layers and improved training algorithms, leading to enhanced performance and efficiency. As a result, these state-of-the-art models such as Mtar (Chandra et al., 2010), Homotarget (Ahmadi et al., 2013), miRNATIP (Fiannaca et al., 2016), miRTDL (Shuang et al., 2016), Deep MirTar (Wen et al., 2018), and miRaw (Pla et al., 2018) interpret the experimentally verified miRNAs and their targets by analyzing their binding site-specific interactions on seed region. The combinatorial functioning of RNN (recurrent neural network) and CNN (convolutional neural network) results in hybrid deep learning approaches miTar (Gu et al., 2021). The CNNs are utilized to extract features from the miRNA-mRNA duplex specifically the base paring and duplex stability, while LSTMs are designed to model temporal dependencies in sequential data, making them well suited for analyzing time-series data. Nowadays, the LSTM-based architectures are demonstrating miRNA target detection (Talukder et al., 2022).
miRNA-lncRNA interaction prediction
Evidence accumulated over the past decade shows that regulation of gene expression involves the interaction with miRNA and lncRNA. The lncRNA biogenesis led to the modulation of chromatin function (Chu et al., 2011; Isoda et al., 2017; Thatai et al., 2023), cytoplasmic mRNA stability and translation (Cesana et al., 2011), and interfere with the signaling pathway (Zhao et al., 2021). In addition, many lncRNAs bearing miRNA complementary sites for miRNAs enabling them to act as competitive endogenous RNAs or sponges of miRNAs that reduce the availability to the miRNA-mRNA target interaction. The stoichiometric relationship between a potential competitive endogenous lncRNA and miRNA is important for achieving a measurable effect on target-mRNA expression (Statello et al., 2021). For example, the lncRNA H19 is found to modulate let-7 family of miRNAs (Kallen et al., 2013).
Several studies have begun to uncover the interactions between miRNA and lncRNA, and more details about the influence of miRNA on lncRNA function are coming into view. Despite extensive research by the exposure of miRNA-lncRNA databases DIANA-LncBase v3 (Karagkouni et al., 2020), LncCeRBase (Pian et al., 2018), and LncRNADisease 2.0 (Bao et al., 2019), the ability to predict miRNA-lncRNA interactions using computational algorithms still needs to be improved by the complexity of the abundance of false-positive results. Resolving the false results leads to several ML tools, listed in Table 5.
MicroRNA-Long Noncoding RNA Interaction Target Prediction Tools
lncRNA, long noncoding RNA.
The upscale growth of neural networks achieved a great success rate in the miRNA lncRNA prediction. A substantial increase of addressing the common methodology, sequence similarity matrix involves incorporation of various kinds of neural network layers for miRNA-lncRNA prediction. The complementary binding strategy of miRNA on lncRNA reflects the sequence similarity matrix as a popular conception in the computational aspects that relies on the seed region. Conservation of lncRNA sequences between different species suggests that they may have important functional roles, and identifying conserved miRNA binding sites within these sequences can provide further evidence for their functional relevance.
A deep convolutional neural network approach was called lncMirNet (Yang et al., 2020b), witnessing k-mer RNA sequence and distributed representation feature of RNA sequence by doc2vec with an accuracy of 85%. The ability of this technique has been adapted to represent RNA sequences as well where the sequence acts as a document and the individual context-dependent meanings of nucleotides or k-mers are treated as words. For SLNPM (Zhang et al., 2019b), sequence similarity and interaction profile similarity by giving mathematical notations for lncRNA interaction profile, lncRNA sequence similarity, and lncRNA interaction profile similarity (k-mer feature). It shows a 99% accuracy.
Bayesian collaborative filtering model called GBCF to pick up ranking list for an individual miRNA or lncRNA based on the known miRNA-lncRNA interactions in the lncRNASNP (Gong et al., 2015) database and it gives sequence, expression, and biological function-based similarity level. The LMI-INGI (Zhang et al., 2021) interactome network and graphlet interaction by sequence similarity matrix regularized the Smith–Waterman algorithm. GKLOLMI link prediction model for inferring miRNA-lncRNA interactions by Gaussian kernel-based method on network profile and linear optimization algorithm by constructing RNA sequence similarity.
miRNA-circRNA interaction prediction
circRNAs, a vast category of noncoding RNAs that are generated through a noncanonical splicing mechanism called backsplicing (Kristensen et al., 2019; Zhang et al., 2013). The evolutionary evidences say that more than 40 years ago, circRNA molecules were discovered in Viroids (Sanger et al., 1976). A few years later, circRNAs were observed in the cytoplasmic fractions of eukaryotic cell lines by electron microscopy (Hsu and Coca-Prados, 1979). Recent studies have highlighted the importance of circRNAs in a variety of biological processes, including development, aging, and disease. Some circRNAs are highly abundant and evolutionarily conserved, suggesting that they may play critical roles in fundamental cellular processes. Furthermore, circRNAs can act as miRNA or protein inhibitors, regulate protein function, or even be translated themselves, highlighting their potential as targets for therapeutic intervention (Memczak et al., 2013; Xia et al., 2017).
The emergence of circBase (Glazar et al., 2014), circRNADb (Chen et al., 2016a), Circad (Rophina et al., 2020), CIRCpedia v2 (Dong et al., 2018), and CircR2Cancer (Lan et al., 2020) databases put forth a standardized information on circRNA. This leads to the information on the interactive association of circRNA with miRNA, and further discovers a biological scenario, listed in Table 6. The construction of circRNA-miRNA interaction matrix on the basis of similarity and associations describes the methodological part of the ML approaches. Most of the recent approaches are built on the neural network-based methods such as WSCD (Guo et al., 2022), CMIVGSD (Qian et al., 2021), GCNCMI (He et al., 2022), SGCNCMI (Yu et al., 2022a), KGDCMI (Wang et al., 2022b), and NGCICM (Ma et al., 2023). These follow an interaction profile kernel similarity network association of circRNA-miRNA.
MicroRNA-Circular RNA Interaction Target Prediction Tools
circRNA, circular RNA.
Each row of the matrix represents a circRNA, while each column represents an miRNA. A high value in a particular cell suggests a strong probability of interaction between the circRNA and miRNA. For example, the GCNCMI, the interaction graph, contains the higher order interaction information of circRNA and miRNA from which we can mine deep semantic information that carries a collaborative signal (He et al., 2022).
The latest one NGCICM incorporates node2vec and graph attention network, conditional random field layer, and inductive matrix completion, together helping to attain an interaction score. The NGCICM model takes the sequence and expression data of circRNAs and miRNAs as input and uses the above techniques to learn a graph representation of the interactions between circRNAs and miRNAs. The model then makes predictions of new circRNA-miRNA interactions based on this learned representation. According to the authors, NGCICM achieves state-of-the-art performance on several benchmark data sets, with an AUC score of up to 0.982, indicating its high prediction accuracy (Ma et al., 2023). Rest of the two methods are based on the network embedding methods NECMA (Lan et al., 2021) and IIMCCMA (Yao et al., 2022).
Network embedding has gained significant attention in recent years due to its ability to learn low-dimensional representations of complex networks, which can improve efficiency, both these models have a higher accuracy more than 90%.
miRNA and mRNA expression profiling
The difficulty in identifying the tissues in which the level of miRNA is expressed is because great limitations exist in the current expression profile detecting techniques, such as microarray (Bargaje et al., 2010). Because the amount of miRNAs contained in a microarray chip is relatively limited, compared with the total amount of miRNAs in the human genome (Nersisyan et al., 2020), this results in establishing an SVM-based approach called Cepred (Wang et al., 2009a) for predicting the coexpression patterns of the human intronic miRNAs with their host genes. Structural characteristics such as distance from the transcription start position of the host gene to the start point of the host intron and miRNA length of the host intron and miRNA act as the features.
The progress of miRNA-mRNA expression correlation databases such as mirCoX (Giles et al., 2013) and DIANA-miTED (Kavakiotis et al., 2022) lead to the comprehensive analysis of expression profiling. In addition, a new method of predicting the expression patterns with the help of linear regression and relevance vector machine from its promoter sequence was introduced by Oğul and Tuncer (2016). ML-based tool maTE (Yousef et al., 2019) integrates differential gene expression data with miRNA target genes. Integrating miRNA regulation with expression data yields powerful results. It is independent of external labels and training data. Another one, the miRcorrNet (Yousef et al., 2021), combined feature grouping and ranking methods to analyze miRNA and mRNA gene expression profiles.
The applicability of this tool in various biological contexts identifies correlation patterns of significant genes linked with a specific miRNA. The predictive model uses ML algorithms, including random forests, gradient boosting, and SVM. The tools and approaches are listed in Table 7.
MicroRNA-mRNA Expression Profiling
miRNA regulatory module detection
MiRNA regulatory modules are groups of miRNAs whose target genes are coregulated and function together in a biological pathway or process. Identifying miRNA regulatory modules helps decipher the evidence of miRNA and mRNA in gene regulatory networks (Masud Karim et al., 2016). ML methods involve analyzing miRNA and gene expression data and identifying coregulated miRNA gene pairs using miRNA-mRNA target interaction. For example, the clustering miRNA-target interactions is a convolutional autoencoder and affinity propagation-based approach for identifying miRNA regulatory modules by extracting miRNA-target interaction features and their overlapping characteristics from the expression profile data (Yang and Wan, 2020). A similar way of prediction by obtaining a reliable relationship between miRNAs and target genes is DeMosa, and it also has the combined effect of stacked autoencoders and K-means clustering (Yang and Song, 2019).
The deep neural network using the miRModuleNet (Yousef et al., 2022) framework has been applied to identify miRNA-mRNA regulatory modules in various biological processes and diseases. For example, miRModuleNet was used to identify miRNA-mRNA regulatory modules involved in breast cancer progression. The identified miRNA-mRNA regulatory modules were enriched in cell cycle regulation and apoptosis pathways, suggesting their potential roles in breast cancer development. An overview of these algorithms or approaches is summarized in Table 8.
MicroRNA Regulatory Module Detection Tools
miRNA disease association prediction
Root causes of human disease development are traced back to miRNA functions in gene expression. Abnormal expression and dysregulations of the miRNA can modulate the metabolic/signaling pathway, which can lead to imbalances in homeostasis. Various bacterial, viral, and protozoan infections can lead to dysregulation of miRNA that significantly affects the signaling pathways influencing the disease outcomes (Agbu and Carthew, 2021; Antil et al., 2022; Ramakrishnan et al., 2023; Ramesh et al., 2023). Understanding disease mechanisms at the miRNA level uncovers the disease biomarkers for diagnosis, medication, prognosis, and prevention (Condrat et al., 2020). For miRNA disease association prediction, the miRNA-disease sequence information and features were majorly taken from searchable databases such as HMDD (Huang et al., 2019b), miR2disease (Jiang et al., 2009), and dbDEMC (Yang et al., 2017).
Data sets having reliable features contribute precision to the output. Prior association of labeled information for investigated miRNAs is required when training an ML model. Most of the approaches such as Jiang et al. (2013), RWRMDA (Chen et al., 2012), EDTMDA (Chen et al., 2019b), and MLMDA (Zheng et al., 2019a) are majorly focused on the functional similarity score information, constructed by integrating miRNA-miRNA interaction data and functional annotation typically depicted as a graph where nodes represent miRNAs and edges represent the functional similarity between miRNAs based on their target genes. Several methods can generate functional similarity scores, including semantic similarity, topology-based similarity, and information content-based similarity. Semantic similarity measures the similarity between miRNA and disease annotations based on their shared ancestor terms in a hierarchical ontology. Topology is based on the number of shared terms and their positions in the ontology hierarchy.
Information content similarity considers the frequency of occurrence of the shared terms in the ontology. Once functional similarity scores are calculated, they can be used to prioritize potential miRNA disease associations. For example, a high functional similarity score between an miRNA and a disease indicates that the miRNA may play a critical role in the pathogenesis of the disease and is likely to be a good candidate for further experimental validation. The heterogeneous graph inference for miRNA-disease association prediction (HGIMDA) (Chen et al., 2016b) is a random walk approach that predicts the association by integrating miRNA functional similarity, disease semantic similarity, Gaussian interaction profile kernel similarity and experimentally verified miRNA-disease associations into a heterogeneous graph. The HGIMDA was applied to human cancers for performance evaluation. An overview of these algorithms or approaches is summarized in Table 9.
MicroRNA-Disease Association Prediction Tools
Predicting the essentiality of miRNA
Short noncoding RNA molecules known as miRNAs play a crucial role in metabolic processes by posttranscriptionally controlling gene expression. Identifying the important miRNAs in the miRNAome requires predicting their essentiality; however, the bioinformatic techniques available for this purpose are limited. Here, a novel approach called miES (Song et al., 2019) is used to rank the significance of miRNAs. The biological features used in miES include the base pair content in precursor and mature miRNAs, mature and nonmature miRNA lengths, and the secondary structure's thermodynamic properties. The complex and deeper features related to miRNAs improve the essentiality prediction. The PESM (Yan et al., 2020) predicts the miRNA essentiality based on gradient boosting machines and miRNA sequences. Compared with miES, PESM integrates more sequences and structural features of miRNAs such as the base pairing propensity and dinucleotide frequency information of precursor and mature miRNAs.
The list of tools and approaches is summarized in Table 10.
MicroRNA Essentiality Prediction Tools
Conclusions
ML has emerged as a valuable component in the multifaceted analysis of miRNAs. The number of novel miRNA features used for training has exploded with data derived from NGS platforms. Prediction of miRNA function had been most explored computationally based on target prediction. These technological advancements have been instrumental in predicting miRNA targets, analysis of their molecular regulatory networks, investigating concepts of miRNA delivery, and studying epigenetic regulation. These are expected to lead to new insights into miRNA function, as well as the development of miRNA-based therapies and diagnostic tools for a wide range of diseases. ML, most recently deep learning, is widely used, and over time, single methods are combined with integrated platforms to improve the plausibility of the predictions. Although novel miRNA biomarkers are being explored from manually curated information, several pipeline tools have also been developed to analyze miRNA-disease associations.
Data integration is becoming critical as miRNA-associated high-throughput sequencing data grow exponentially.
The most troubling aspects of the current trends were the tool updations or follow-ups and availability of active tools. Most studies are approaches, especially disease association studies, because developing an approach to a tool requires more compelling data, computing efficiency, and accurate maintenance. However, many early tools are still well maintained and frequently updated or integrated with new tools. Predictably, future miRNA ML tools will contain the following characteristics: aim for new miRNA knowledge, analyze high-throughput miRNA technology data, integrate multilevel omics data, and focus on human disease. These collectively highlight the key trends in miRNA ML tool development, researchers can ensure that future miRNA tools are better equipped to meet the needs of the scientific community and address important questions related to miRNA integrative biology.
Key Points and Executive Highlights
The miRNA biogenesis is critical to the posttranscriptional regulation and the emergence of miRNA-based databases diversified the miRNA biology to be visualized at various layers for ML-based applications.
ML approaches have great potential to contribute to the miRNA regulatory network of mRNA, lncRNA, and circRNAs.
Artificial neural network, SVM, and random forest marks a major part of ML in miRNA research.
The lack of availability of reliable tools pertaining to miRNA promoter prediction, target prediction, precursor classification, and disease association is a significant concern in the field.
Development of efficient and well-maintained tools is necessary to effectively extract both sequence and structural features of miRNA from different database resources.
Taken together, this review critically examines and unpacks the cutting-edge synergy of ML approaches and miRNA research so as to develop a dynamic and microlevel understanding of human health and diseases.
Footnotes
Acknowledgment
The authors wish to thank Dr. Ajith V. Pankajam, National Cancer Institute, National Institutes of Health (NIH), USA, for critical comments and feedback and an earlier version of the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.