
Abstract
Imatinib (IM), a breakthrough in chronic myeloid leukemia (CML) treatment, is accompanied by discontinuation challenges owing to drug intolerance. Although BCR-ABL1 mutation is a key cause of CML resistance, understanding mechanisms independent of BCR-ABL1 is also important. This study investigated the sphingosine-1-phosphate (S1P) signaling-associated genes (SphK1 and S1PRs) and their role in BCR-ABL1-independent resistant CML, an area currently lacking investigation. Through comprehensive transcriptomic analysis of IM-sensitive and IM-resistant CML groups, we identified the differentially expressed genes and found a notable upregulation of SphK1, S1PR2, and S1PR5 in IM-resistant CML. Functional annotation revealed their roles in critical cellular processes such as proliferation and GPCR activity. Their network analysis uncovered significant clusters, emphasizing the interconnectedness of the S1P signaling genes. Further, we identified interactors such as BIRC3, TRAF6, and SRC genes, with potential implications for IM resistance. Additionally, receiver operator characteristic curve analysis suggested these genes’ potential as biomarkers for predicting IM resistance. Network pharmacology analysis identified six herbal compounds—ampelopsin, ellagic acid, colchicine, epigallocatechin-3-gallate, cucurbitacin B, and evodin—as potential drug candidates targeting the S1P signaling genes. In summary, this study contributes to efforts to better understand the molecular mechanisms underlying BCR-ABL1-independent CML resistance. Moreover, the S1P signaling genes are promising therapeutic targets and plausible new innovation avenues to combat IM resistance in cancer clinical care in the future.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative malignancy characterized by a chromosomal rearrangement event of genes resulting in the BCR-ABL1 fusion gene. This genetic change is known as the Philadelphia chromosome, induces continual activation of tyrosine kinase, and thereby, leads to irregular cellular signaling and proliferation, contributing to CML progression (Li et al., 2022; Zaker et al., 2023). CML treatment involves tyrosine kinase inhibitors (TKIs), mainly imatinib (IM), as the first-line therapy. After the introduction of IM, the survival rate of patients has improved, and yet, 20–30% of the patients develop resistance over time owing to intolerance to the drug (Xu et al., 2023).
CML resistance is usually caused by BCR-ABL1 gene alterations. However, some patients may also develop BCR-ABL1-independent resistance owing to epigenetic alterations, alternative signaling pathway survival, drug efflux mechanisms, autophagy, and antiapoptotic pathway activation (Alves et al., 2021; Meenakshi Sundaram et al., 2019). Unraveling the exact etiologies behind BCR-ABL1-independent resistance remains an ongoing challenge.
Recently, sphingosine-1-phosphate (S1P) signaling pathway has been widely recognized owing to its pivotal role in disease progressions, including cancer. This pathway involves the conversion of sphingosine to S1P, a lipid mediator phosphorylated by sphingosine kinase 1 (SphK1). After being produced, S1P interacts with G protein-coupled S1P receptors (S1PR1–S1PR5) and then triggers downstream signaling to develop diseases by regulating cell growth, survival, and inflammation (Chen et al., 2022a; Maceyka et al., 2012; Proia and Hla, 2015).
The importance of the S1P signaling pathway in cancer biology has been well investigated (Pyne and Pyne, 2020; Zheng et al., 2019). Both clinical and in vitro research have confirmed the link between its involvement and patients’ poor prognosis (Pyne et al., 2012; Tsuchida et al., 2017; Wang et al., 2019; Yu et al., 2021). Additionally, a meta-analysis conducted by Zhang et al., 2022 also documented the unfavorable prognosis of SphK1 expression in patients with solid tumors (Zhang et al., 2022). Several studies also indicate that this pathway has been implicated in driving drug resistance in cancer, including hematological malignancies (Bonhoure et al., 2006; Fu et al., 2017; Matula et al., 2015; Ren and Su, 2020; Rosa et al., 2013).
The existing studies on resistant CML associated with BCR-ABL1-dependent mechanism also indicate the influence of elevated S1P signaling genes, i.e., SphK1 and S1PRs in this context (Baran et al., 2007; Marfe et al., 2011; Salas et al., 2011). However, the involvement of this pathway in resistant CML independent of the BCR-ABL1 mechanism is unclear, suggesting more research. Therefore, exploring this pathway may reveal therapeutic options and resistance mechanisms.
In the present study, we used a multiomics approach to investigate the role of S1P signaling-associated genes driving IM resistance in CML through the BCR-ABL1-independent mechanism. In IM-resistant CML, transcriptomic data analysis revealed upregulated S1P signaling genes. A network pharmacology technique was also used to identify herbal drug candidates that can target S1P signaling genes, thereby expanding our therapeutic options. The study workflow is shown in Figure 1.
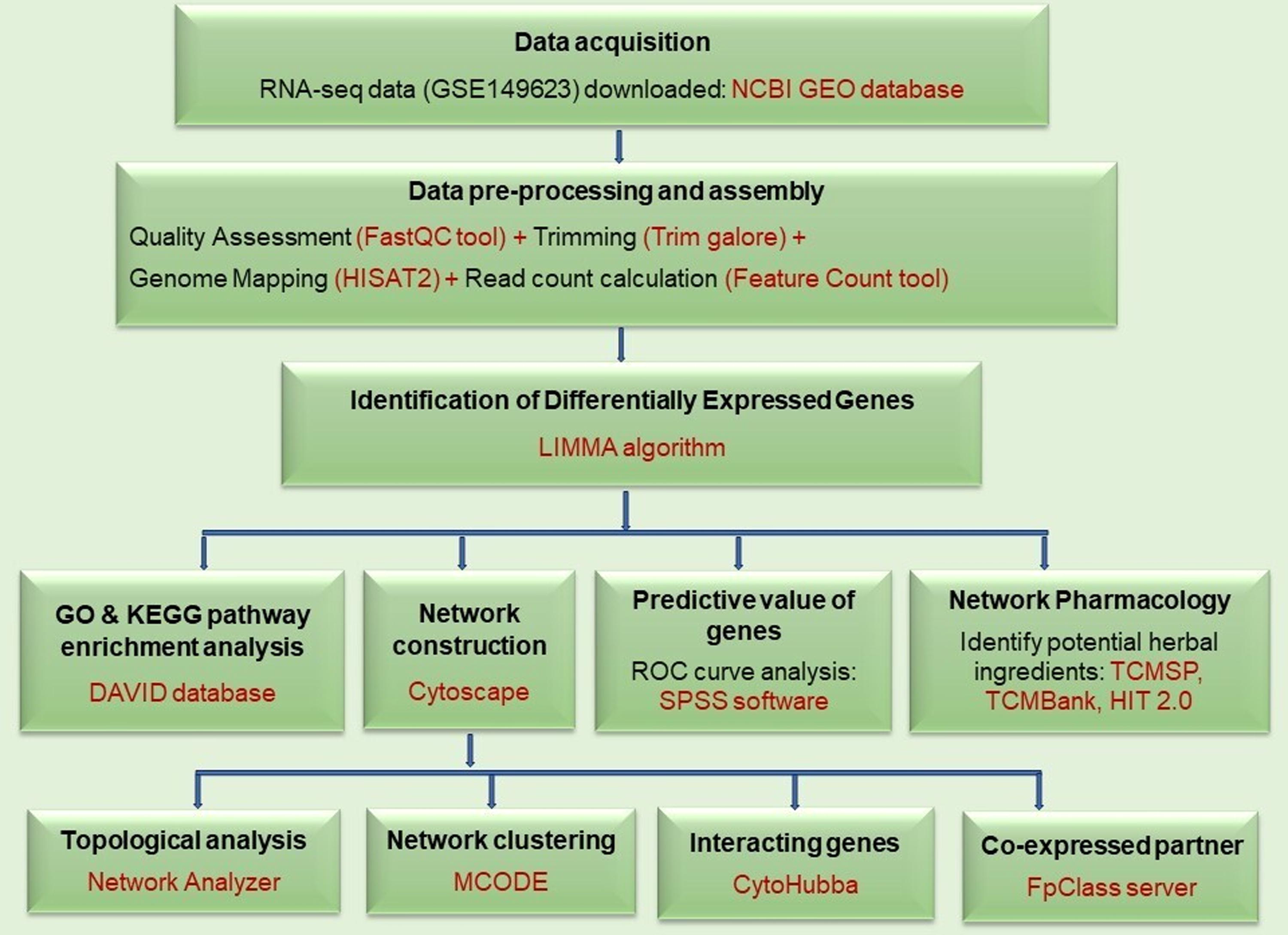
Workflow for the study.
Materials and Methods
The present study was conducted with the use of publicly available data and conducted under the overall research ethics oversight of the authors’ institutions.
Dataset collection and inclusion criteria
The RNA-seq datasets for IM-resistant and sensitive CML were sought in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home) databases using “Imatinib Resistant” and “Chronic Myeloid Leukemia” as search terms. Subsequently, dataset GSE149623 (Project accession number PRJNA629413) was included in our study based on specific criteria: (i) containing both IM-resistant and IM-sensitive CML group; (ii) demonstrating resistance through BCR-ABL1-independent mechanisms; (iii) utilizing clinical samples or cell lines; and (iv) having paired-end reads.
Data preprocessing and assembly
RNA-seq data in FASTQ format were first uploaded onto the Galaxy web server (https://usegalaxy.org/) and underwent quality assessment before and after the preprocessing step using the FASTQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). This preprocessing involved adaptor trimming and removal of low-quality bases from the reads utilizing Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), selectively retaining reads with a phred-score of ≥30, and the validated reads were then mapped to the human genome (GRCh38/hg38) using the HISAT2 aligner to generate BAM files (Kim et al., 2015). Then, the quantification of aligned reads from these BAM files was executed using the FeatureCount tool (Liao et al., 2014).
Differentially expressed genes (DEG) analysis
DEGs between IM-resistant and IM-sensitive CML were identified using the LIMMA package of R (Ritchie et al., 2015). An adjusted p-value <0.05 and | log2 FC (fold change)| >0 were considered as cut-off parameters. Notably, genes of interest, such as S1P signaling-associated genes in the resistant CML group were specifically singled out for subsequent in-depth analysis.
Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis
Biological processes (BPs), cellular components (CCs), molecular functions (MFs), and pathways were discovered using GO and KEGG pathway enrichment analysis, facilitated by the Database for Annotation, Visualization and Integrated Discovery (https://david.ncifcrf.gov/) and visualized by SR plot (https://www.bioinformatics.com.cn/en). Significant enrichment of functional annotations and pathways was determined based on a threshold of p-value <0.05.
Construction of S1P signaling-associated genes network
Upregulated DEGs were initially imported into the Cytoscape (v3.10.1) visualization software (Shannon et al., 2003). Subsequently, the stringApp (v2.0.1) plugin was utilized within Cytoscape to establish a protein–protein interaction (PPI) network, where proteins (or genes) acted as nodes interconnected by edges reflecting protein interactions (Szklarczyk et al., 2023). Following this, the Analyze Network tool, an embedded plug-in within Cytoscape, was employed to compute the network’s topological attributes for an in-depth analysis of its characteristics (Assenov et al., 2008). These networks were then clustered using the MCODE (chemical complex detection) plug-in with default parameters (degree cut-off = 2, node score cut-off = 0.2, K-core = 2, and max. depth = 100) to identify relevant molecular modules within the network (Bader and Hogue, 2003). Furthermore, using the CytoHubba plug-in, genes that interact with our genes of interest (SphK1, S1PR2, and S1PR5) were identified (Chin et al., 2014). Finally, these identified genes as co-expression partners of SphK1, S1PR2, and S1PR5 were validated by the FpClass server (http://dcv.uhnres.utoronto.ca/FPCLASS/ppis/), indicating a consistent pattern of correlation or connected expression levels across different situations (Kotlyar, 2014).
Statistical analysis
The statistical significance of group comparison was determined using the independent t-test. SPSS software version 26.0 (IBM Corp., Armonk, NY) was utilized for the statistical analysis and a significance level of p < 0.05 was considered.
The receiver operator characteristic (ROC) curve analysis was employed to ascertain the predictive capacity of SphK1, S1PR2, and S1PR5 genes in anticipating IM resistance. With this ROC, an area under the curve (AUC) was found, which functioned as a quantitative assessment to measure the efficacy of these genes as potential biomarkers across the sample population.
Network pharmacology
A network pharmacology approach was employed to identify potential herbal bioactive ingredients (or compounds) that can influence target gene activity. Ingredients were identified by using several databases, including Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) (https://tcmsp-e.com/tcmsp.php), Traditional Chinese Medicine (TCM) Bank (https://tcmbank.cn/), and Herbal Ingredients’ Targets Platform (HIT2.0) (http://hit2.badd-cao.net/).
The TCMSP and TCM Bank databases provide information related to traditional Chinese medicine that shows relationships between herbs, ingredients, targets, and associated diseases. Information on various pharmacokinetic properties, including oral bioavailability (OB), drug-likeness (DL), blood–brain barrier, intestinal epithelial permeability, molecular interactions, and other chemical properties can also be explored. HIT2.0 also provides herbal ingredients and targets data but it is based on literature evidence.
Screening of bioactive ingredients
By entering the target gene names (SphK1, S1PR2, and S1PR5) on TCMSP, TCMBank, and HIT2.0 databases, potential ingredients were identified. These ingredients were then evaluated for the TCMSP database’s criteria: OB ≥ 20% and DL ≥ 0.18. These two parameters are crucial for screening potential drug candidates as well as in evaluating the absorption, distribution, metabolism, and excretion properties of the drugs. Drug design employs qualitative pharmacokinetics to enhance drug properties, including solubility and chemical stability, with a DL value ≥0.18. OB is another important pharmacokinetic parameter since drug distribution in the body requires a minimum OB value of 20%.
Herbs associated with these identified ingredients and targets were also extracted. We then used Cytoscape (v3.10.1) software to construct and visualize a network that links targets, bioactive ingredients, and herbs.
Toxicity prediction of bioactive ingredients
ProTox 3.0 (https://tox.charite.de/protox3/) server was used to predict the toxicity of the identified herbal ingredients. Information on acute, organ, and endpoint toxicity was extracted by inputting the compound’s (or ingredient’s) SMILE format. Along with acute and chronic toxicity prediction, this server also predicts molecular initiating events, metabolism, nuclear receptor signaling, and stress response pathways.
Results
DEG analysis and S1P signaling genes
The study included an RNA-seq dataset comprising three IM-resistant and three IM-sensitive cell lines. RNA-seq data analysis revealed differential expression in 10,420 genes between the two groups, with 4975 genes upregulated and 5445 genes downregulated, respectively (Supplementary Table S1) in the IM-resistant group. DEGs were visually represented as a heatmap (Supplementary Fig. S1). For subsequent analysis, genes associated with the S1P signaling pathway were considered. SphK1, S1PR2, and S1PR5 were identified as upregulated in the IM-resistant group, while S1PR3 exhibited downregulation (Table 1).
The Expression Value of the S1P Signaling Pathway-Associated Genes in Imatinib-Resistant vs Imatinib-Sensitive Group
FC- fold change; S1P- sphingosine-1-phosphate.
GO annotation and KEGG pathway enrichment analysis
Enrichment was studied to determine which BPs and MFs of SphK1, S1PR2, and S1PR5 genes may contribute to CML resistance (Fig. 2A and B). These BPs such as S1P signaling pathways, metabolic activities, cell migration, and proliferation were regulated by our target genes. Their MFs include S1PR activity, lipid binding, and G protein-coupled receptors. In terms of CC, these genes were primarily linked with the plasma membrane and cytoplasm. The KEGG pathway analysis revealed a link with the S1P signaling pathway. This enrichment sheds light on how these genes may regulate cellular activities and structure, hence, contributing to resistance mechanisms.

Enrichment analysis of the S1P signaling-associated genes: SphK1, S1PR2, and S1PR5.
Construction of S1P signaling-associated genes network
To investigate the functional implications of resistant DEGs (upregulated) at the protein level, a PPI network was constructed. This network comprised 4314 nodes and 70,096 edges after filtering genes with a medium confidence score >0.4.
Topological attribute analysis
Assessing key topological attributes like network diameter, clustering coefficient (Ci), characteristic path length, degree, betweenness centrality (BC), and closeness centrality revealed protein characteristics and functions within the network, emphasizing their biological significance (Supplementary Table S2).
The network diameter is the mean distance of the shortest paths between any two nodes, which provides insights into the network’s architecture, interconnectivity, and interaction capabilities with other nodes. Understanding diameter aids in figuring out how efficient, robust, and connected the network is, thereby making it a crucial topological attribute (Clarke et al., 2000). Ci is the tendency to cluster networks. Since Ci values range from 0 to 1, cluster formation increases as Ci approaches 1 (Koutrouli et al., 2020). Our analysis found a moderate cluster formation rate of 0.275. Additionally, the network of our study revealed a significant biological pattern, because of its wider diameter and lesser density.
Characteristic path length (average path length, or APL) measures a network’s navigability by averaging the shortest distance between all the nodes, as well as its proximity and speed of information flow. A shorter APL can transfer signals faster, help in drug target identification, and provide drug activity information (Asif et al., 2014; Xu et al., 2011). In our study, the APL was found to be large and its Ci was small, suggesting that it was not constrained by scale. This scale-free attribute suggested that most nodes (proteins or genes) within the network had a few connections, while certain nodes (hubs) were linked to numerous nodes, a prevalent attribute of scale-free networks.
The degree of a node indicates the number of connections it has to other nodes. BC determines each node’s importance within a network and is calculated by dividing the total number of shortest paths passing through the node by the sum of all these shortest paths. Evidence suggests that high BC proteins play a key role in networks’ modularization. On the contrary, closeness centrality identifies fast-communicating network nodes. Finding the key metabolites in a biochemical network is a common application of closeness centrality (Koutrouli et al., 2020; Zhao and Liu, 2019). In our study, we assessed the degree, BC, and closeness centrality of our target genes (SphK1, S1PR2, and S1PR5) (Supplementary Table S2). These centrality parameters revealed our target gene connections, BC showed which genes were important for our targets, and closeness centrality showed which genes communicated fast with our target genes.
Cluster analysis
Network cluster analysis showed 92 modules. S1P signaling genes were in module 38, which had 21 nodes and 33 edges (Fig. 3A). This finding indicates a high degree of connectivity and functional relevance between these S1P signaling genes and other nodes in cluster 38 in our network architecture, necessitating further exploration into their combined functional consequences.

Identification of interacting genes and validation of co-expression partners (or genes)
Based on the degree method of CytoHubba, S1P signaling genes interacting with other genes were assessed (Fig. 3B). The gene co-expression score, which was derived from Pearson correlation of expression profiles, and the network topology score indicating gene presence in training data and interaction strength were used to validate co-expressed genes of SphK1, S1PR2, and S1PR5 (Supplementary Table S3). Our analysis identified that BIRC3, PRKCZ, TRAF6, CALM3, CTNNB1, and SRC were co-expression partners of SphK1 and GNA13 co-expressed with S1PR2. However, the inconclusive confirmation was caused by the fact that the co-expressed genes for S1PR5 were different.
Upon analyzing the ROC curve conducted on both the IM-resistant and IM-sensitive datasets, it was observed that all three S1P signaling genes exhibited AUC values exceeding 0.9 (Supplementary Fig. S2). This indicates a high level of predictive accuracy or discriminatory power of these genes in distinguishing between IM-resistant and IM-sensitive cases.
Network pharmacology
Identification of potential bioactive ingredients
Six ingredients—ampelopsin, ellagic acid, colchicine, epigallocatechin-3-gallate (EGCG), cucurbitacin B, and evodin—met the predefined criteria (OB ≥ 20% and DL ≥ 0.18) and were therefore identified as potential drug candidates (Supplementary Table S4). We also investigated the herbal sources linked to these ingredients and their targets, which revealed significant associations among them. Ampelopsin and ellagic acid were obtained from Morus alba herb; ampelopsin and EGCG were obtained from Ginkgo biloba seed, while ellagic acid and EGCG were sourced from Eriobotryae folium.
Target-bioactive ingredient-herb network
The constructed network of target-ingredient-herb associations comprised 111 nodes and 116 edges (Fig. 4). Among these nodes, three were gene targets, six were ingredients, and the remaining nodes represented herbs. Among the gene target nodes, S1PR2 exhibited the highest degree value (=4), indicating its interaction with four ingredients. This emphasizes S1PR2’s importance and centrality in the network, indicating its vital role in network connectivity and interaction.
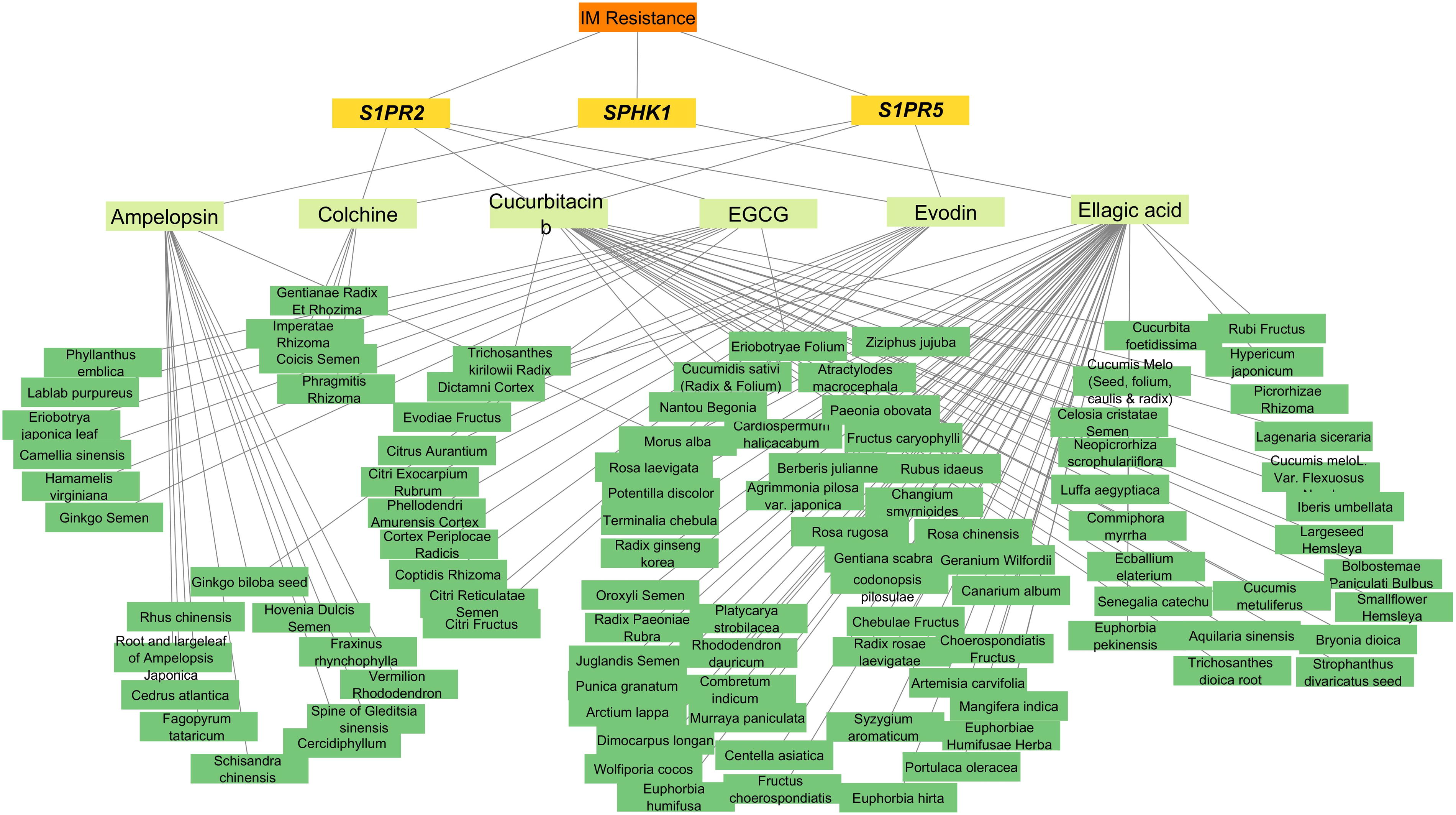
Target-ingredient-herb network.
Toxicity prediction of bioactive ingredients
Four ingredients have shown no acute toxicity, but cucurbitacin B and evodin may be somewhat and moderately harmful and irritating. In hepatotoxicity, long-term ellagic acid consumption can damage the liver, while the other five ingredients are harmless. Cardiotoxicity analysis shows only evodin is active while other ingredients are harmless. Except for ampelopsin, five components have demonstrated no mutagenic potential. Ampelopsin and cucurbitacin B may be carcinogenic. A detailed description of the ingredients' toxicity predictions can be found in Supplementary Table S5.
Discussion
The molecular basis of BCR-ABL1-independent IM resistance in CML is still inadequately understood. BCR-ABL1-independent IM resistance is relevant for CML clinical care as a primary or secondary contributor to treatment resistance (Patel et al., 2017). Therefore, an effort was made in the present study so as to understand the alternative pathways for developing effective therapeutic strategies for TKI-resistant CML patients associated with the BCR-ABL1-independent mechanism.
We aimed to uncover the genes associated with the S1P signaling pathway by integrating transcriptomics and a network pharmacology approach in relation to their roles in IM resistance. Transcriptomic data analysis showed that IM-resistant CML cell lines have increased SphK1, S1PR2, and S1PR5 genes. This supports other studies linking S1P signaling genes to BCR-ABL1-dependent IM resistance (Baran et al., 2007; Marfe et al., 2011; Salas et al., 2011). Beyond CML, SphK1 overexpression conferred resistance to chemotherapeutic drugs in acute myeloid leukemia (AML) and UV irradiation in gastric and prostate cancer cells (Bonhoure et al., 2006; Pchejetski et al., 2005; Xiong et al., 2014; Zhang et al., 2021). Zhang et al. (2021) found that S1P causes S1PR2 uptake in colorectal cancer, causing 5-fluorouracil (5-FU) resistance. According to animals and cell lines investigations, S1PR2 enhances autophagy-mediated uracil synthesis, decreasing 5-FU efficacy (Zhang et al., 2021). Fu et al. (2017) also found S1PR1, S1PR2, and S1PR5 genes in resistant myeloma cells, with S1PR5 being the most expressed gene while S1PR3 and S1PR4 were undetectable. This finding is consistent with our investigation. Collectively, all these findings conclude that S1P signaling genes induce resistance to therapeutics across diverse cancer types.
The GO enrichment analysis conducted in this study sheds light on the diverse BPs and MFs associated with S1P signaling genes. The role of SphK1, S1PR2, and S1PR5 genes in migration, proliferation, and metabolic control could provide a mechanistic understanding of these genes in IM resistance. Fu et al. (2017) revealed how S1P signaling genes affect myeloma cell viability and treatment resistance. The study discovered that the S1PR1–S1PR5 genes turned on downstream signaling genes. These genes then turned on the PI3K/Akt/mTOR and STAT3/bcl-2 pathways, which helped cells grow (Fu et al., 2017). The SphK1/S1P pathway also reports cell proliferation, survival, and apoptosis prevention in chronic lymphocytic leukemia (CLL) and AML (Almejún et al., 2017; Powell et al., 2017). Hence, parallel results from different studies show how important S1P signaling genes are for resistance in several different types of hematological malignancy.
The PPI network unveiled the complex relationships among S1P signaling genes, emphasizing their interconnectedness. Cluster analysis revealed significant molecular modules, offering a systems-level view of how these genes collaboratively contribute to the resistant phenotype. With confidence levels from medium (above 0.4) to highest (above 0.9), the PPI network built with STRING provides both direct and indirect correlations (Szklarczyk et al., 2023). We included interactions with strong evidence, used a confidence level of 0.4 in our study, and examined the validity of these links.
Finding and confirming co-expression genes (BIRC3, PRKCZ, TRAF6, CALM3, CTNNB1, SRC, and GNA13) adds to our knowledge and shows how S1P signaling genes work together in the setting of drug resistance. Elevated SphK1 expression is reported to inhibit apoptosis induced by doxorubicin or docetaxel in non-small cell lung cancer. This resistance is linked to the elevated level of antiapoptotic proteins (IAPs), including BIRC3 (also known as c-IAP2) (Song et al., 2011). Hematological malignancies also exhibit elevated cIAP2 expression, contributing to adult T-cell leukemia and CLL (Fulda, 2014). Furthermore, the c-IAP2-MALT1 (mucosa-associated lymphoid tissue) hybrid protein also contributes to B-cell malignancy and MALT lymphoma by persistently activating aberrant NF-kB transcription factor (Varfolomeev et al., 2006).
Aberrant activity of SRC (sarcoma proto-oncogene kinase) has been associated with the activation of SphK1 in glioblastoma and estrogen receptor-negative breast cancer (Alshaker et al., 2014). Despite the lack of a direct association between SRC and S1PR2 gene in co-expression analysis, our CytoHubba analysis revealed an association, consistent with prior findings. This correlation is further supported by findings from breast cancer cell line studies and an animal model of neuroinflammation (Ohotski, 2014; Sancho-Alonso et al., 2023).
GNA13 protein is known for its role in regulating tumor behavior across diverse hematological malignancies, including diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (Dubois et al., 2016; Shimono et al., 2018). Existing findings have elucidated a connection between GNA13 and S1PR2 signaling, highlighting their significance in the regulation of proliferation in both germinal center B-cells lymphoma and DLBCL (Muppidi et al., 2014; Stelling et al., 2018).
There is a growing interest in the use of herbal bioactive ingredients for the treatment of cancer owing to the evidence that ingredients like curcumin, resveratrol, EGCG, and berberine exhibit anticancer properties by influencing drug resistance, autophagy, and apoptosis pathways, regulating the tumor microenvironment, exerting cytotoxic action, balancing immunity, and enhancing chemotherapy in both in vitro and in vivo settings (Ali et al., 2023; Chen et al., 2022b; Luo et al., 2019). Unfortunately, owing to their inadequate bioavailability, herbal ingredients have limited clinical value and can be improved by employing a nanotechnology-based drug delivery system (Ali et al., 2023). Before delving into nanobiotechnology, we suggest that the network pharmacology approach can be used to better understand drug pharmacology by revealing complex interactions and predicting new effects, contributing to the safe and widespread application of herbal ingredients.
The network pharmacology approach systematically investigates and identifies possible herbal compounds, as well as their relationships to target genes. One of the most linked genes in the target-ingredient network is S1PR2, which interacts with colchine, EGCG, cucurbitacin b, and evodin. Ellagic acid has the most linkages in the ingredient-herb network. Drug ADMET characteristics show that ellagic acid has an OB of 43.06 and a DL of 0.43. These findings show that network pharmacology can help us understand therapeutic routes and address herbal ingredient bioavailability concerns in CML treatment.
Ampelopsin (dihydromyricetin) shows potential in fighting against cancer. It lowers the levels of biological factors that support cancer, raises the production of proteins that cause apoptosis, stops tumor growth by controlling proteins that create blood vessels and spread cancer cells, and also lowers the levels of inflammatory markers in cancer cells (Tuli et al., 2022). The major effects of colchicine are tubulin degradation and mitotic metaphase arrest and it also reduces inflammation and innate immunity (Leung et al., 2015). Ellagic acid induces apoptosis, inhibits DNA binding to carcinogens, and impairs tumor formation and metastatic drug resistance (Zhang et al., 2014). EGCG degrades PML/RAR fusion protein to induce apoptosis and decrease cell growth (Qian et al., 2005). Cucurbitacin B inhibits cell proliferation, migration, invasion, apoptosis, and the cell cycle (Cai et al., 2015). Evodin causes apoptosis by producing too much reactive oxygen species, which stops the cell cycle in G2/M (Chen et al., 2021). A detailed mechanism of action of all these six ingredients is provided in Supplementary Table S6. We can investigate these drugs for possible application in imatinib-resistant CML by taking a closer look at the anticancer mechanisms of the herbal components we have found.
Limitations
Firstly, to obtain more robust results, additional research with a larger sample size is required since the sample size in the present study is limited. Secondly, genes involved with BCR-ABL1-independent resistant CML have been identified using cell line data. These should also be verified using clinical samples. Because the intricacies of clinical context might not fully replicate the cell line nature, it is necessary to conduct translational research and clinical studies to ascertain the potential biomarker candidates. Thirdly, to confirm the efficacy of herbal bioactive ingredients, clinical and animal experiments are required.
Conclusions
Our findings suggest that the genes SphK1, S1PR2, and S1PR5 may have a role in the IM resistance in CML, independent of BCR-ABL1 mechanisms. Developing new drugs that modulate these target genes can pave the way for therapeutic innovations in CML. Furthermore, the toxicity assessment of the identified herbal ingredients or drug candidates is poised to inform future translational clinical studies in drug discovery and development. Taken together, these findings advance our understanding of the complex mechanism of IM-resistant CML, independent of BCR-ABL1, and may guide future investigations toward effective therapeutic strategies.
Footnotes
Authors’ Contributions
S.M.: Conceptualization, methodology, data curation, investigation, formal analysis, writing—original draft preparation. M.B.: Conceptualization, methodology, supervision, formal analysis, writing—review and editing. V.U.: Data curation, formal analysis, statistical analysis. S.T.: Supervision, reviewing and editing S.H.: Supervision, reviewing, and editing.
Author Disclosure Statement
The authors declare no potential conflicts of interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.