
Abstract
The study of longevity and its determinants has been revitalized with the rise of microbiome scholarship. The gut microbiota have been established to play essential protective, metabolic, and physiological roles in human health and disease. The gut dysbiosis has been identified as an important factor contributing to the development of multiple diseases. Accordingly, it is reasonable to hypothesize that the gut microbiota of long-living individuals have healthy antiaging-associated gut microbes, which, by extension, might provide specific molecular targets for antiaging treatments and interventions. In the present study, we compared the gut microbiota of Chinese individuals in two different age groups, long-living adults (aged over 90 years) and elderly adults (aged 65–74 years) who were free of major diseases. We found significantly lower relative abundances of bacteria in the genera Sutterella and Megamonas in the long-living individuals. Furthermore, we established that while biological processes such as autophagy (GO:0006914) and telomere maintenance through semiconservative replication (GO:0032201) were enhanced in the long-living group, response to lipopolysaccharide (GO:0032496), nicotinamide adenine dinucleotide oxidation (GO:0006116), and S-adenosyl methionine metabolism (GO:0046500) were weakened. Moreover, the two groups were found to differ with respect to amino acid metabolism. We suggest that these compositional and functional differences in the gut microbiota may potentially be associated with mechanisms that contribute to determining longevity or aging.
Introduction
Aging and its determinants are researched worldwide as with interventions that may prove to have antiaging effects. According to data published by the National Bureau of Statistics, China became an aging society in 2013. Over the 6 years that followed, the number of adults aged 60 and over increased by 51 million, whereas the number of adults aged 65 and over increased by 44 million, thereby revealing that the trend in population aging has been gradually gaining in prominence (Xiang and Wang, 2021). At present, the average life expectancy of a Chinese citizen is 76.4 years, although the average healthy life expectancy is somewhat shorter at 68.7 years, thereby indicating that on average, during the final 8 years of life, China’s elderly population is likely to experience suboptimal health. This is reflected in the rising prevalence of chronic diseases and an increasing number of disabled elderly adults (Wang, 2019), which has placed a heavy burden on caregiving at family, society, and country levels. These trends accordingly highlight the necessity of gaining a better understanding of the factors that influence aging and longevity and the veritable need to develop measures and targeted guidance for promoting healthy aging in China and worldwide.
Human longevity has been established to be influenced by both genetic (e.g., genomic variation and chromosome stability) and environmental (e.g., gut microbiota, diet, exercise, air quality, and living environment) factors. Among these factors, the gut microbiota is considered the most important component of the human microecosystem, the composition and activities of which undergo continuous change with age (Bradley and Haran, 2024). The findings of numerous studies have provided evidence to indicate that the composition and function of the gut microbiota are closely associated with the host health status (Ghosh et al., 2022; Sun and Li, 2021). In all, compared with healthy individuals, those with poor health or elderly adults appear to have reduced levels of gut microbial diversity and increased proportions of pathogenic microorganisms. These differences are not only associated with adverse effects on gastrointestinal digestion and absorption but also are linked to diseases and morbidity in several other physiological systems, including the cardiovascular, immune, neurological, and respiratory systems (Haran and McCormick, 2021).
Insofar as the mechanisms are concerned, gut microbes can influence the development of age-related diseases and senescence by modulating the body’s immune responses and metabolic pathways (Buford, 2017). For example, one of the pathogenic mechanisms associated with age-related diseases is chronic low-grade inflammation (i.e., inflammaging). The expression of pro-inflammatory mediators, including tumor necrosis factor-α and interleukin-6, may be induced during the aging process, which will in turn promote the activation of a number of signaling pathways that can negatively modulate immune function, thereby leading to a progressive deterioration of the immune system, a process referred to as immunosenescence. A majority of age-related diseases and disorders are believed to be associated with inflammaging and immunosenescence processes, the former of which has been established to be accelerated by gut dysbiosis (Haran and McCormick, 2021).
Compared with the general elderly population, it has been found that centenarians, as representatives of a population with longevity, are characterized by significant differences in the composition and function of their gut microbiota. For example, a Japanese study identified specific bacterial strains in the gut microbiota of centenarians that can produce unique secondary bile acids, which regulate immune cells and reduce the risk of pathogenic infections, thereby contributing to the maintenance of intestinal homeostasis (Sato et al., 2021). Similarly, the findings of an Italian study revealed that gut microbes in centenarians (aged 99–104 years) and semi-supercentenarians (aged 105–109 years) were better adapted for the degradation of xenobiotics and were characterized by modifications in pathways associated with carbohydrate, amino acid, and lipid metabolism (Rampelli et al., 2020).
In this study, in which we sought to characterize the gut microbiota of long-living individuals in Quzhou, China, we collected fecal samples from long-living adults (aged over 90 years) and elderly adults (aged 65–74 years) who were free of major diseases. To examine the composition of the gut microbiota in these individuals, we performed metagenomic sequencing and compared the two groups to identify potential gut microbiota signatures of longevity. Moreover, on the basis of functional analysis of the metagenomic sequencing data, we propose microbial mechanisms and pathways that are potentially associated with longevity.
Materials and Methods
Participants and fecal collection
Twenty adults aged over 90 years who had no major acute or chronic diseases (long-living group) and 20 adults aged 65–74 years (elderly group) who had no major acute or chronic diseases as determined by medical history and examination were randomly selected from the respective populations in Kaihua County, Quzhou City. Fecal samples were collected from these individuals using BioYouTM fecal preservation tubes and were stored at −20°C prior to analysis.
This study has been conducted in accordance with the ethical standards according to the Declaration of Helsinki and the national and international guidelines and has been approved by the research ethics committee at the Medical School, Quzhou College of Technology and the People’s Hospital of Kaihua, Quzhou, and a written informed consent was obtained from all study participants.
DNA extraction and next-generation sequencing
The total DNA was extracted from samples using the Fecal Genome DNA Extraction Kit (AU46111-96, BioTeke, China). DNA libraries were constructed using the TruSeq Nano DNA Library Preparation Kit-Set (#FC-121–4001, Illumina, USA) following the manufacturer’s instructions. Metagenome libraries were then sequenced on an Illumina NovaSeq 6000 platform with PE150 at LC-Bio Technology Co., Ltd. (Hangzhou, China).
Sequencing data analysis
Sequencing adapters were removed from demultiplexed raw sequences using cutadapt (v1.9). Then, the low-quality reads (quality scores <20), short reads (<100bp), and reads containing more than 5% “N” records were trimmed using the sliding-window algorithm method in fqtrim (v 0.94). Quality filtered reads were first aligned to human genome using Bowtie (v2.2) to filter out host contaminations. Then, the remaining reads were subjected to de novo assembly for each sample using MEGAHIT (v1.2.9) and used to assign microbial functions and taxonomy. MetaGeneMark (v3.26) was used to predict the coding sequence (CDS) of the assembled contigs, and CDSs of all samples were clustered using CD-HIT (v4.6.1) to obtain unigenes. DIAMOND (v0.9.14) was used to perform a taxonomic assessment of the microbiota based on the nonredundant protein sequence (NR) database.
Statistical analyses
Species richness and evenness were evaluated using selected α-diversity indices, namely, the Chao1, observed species, Good’s coverage, and Shannon indices. The variability of species among samples was determined based on analyses of β-diversity performed using principal component, cluster, similarity, and multivariate analysis of variance analyses. The Mann–Whitney U test was used to examine differences in species abundance, functional composition, and unigene expression between the long-living and elderly individuals based on screening thresholds of |log2(fold_change)|≥1 and p < 0.05. Then the Gene Ontology (GO) (2018.12.21) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (v87.1) enrichment analysis were performed with different expressed unigenes. Finally, machine learning algorithms, such as random forests, were used to identify key species that can be used to discriminate between the two groups.
Results
Composition and diversity of the gut microbiota
The raw metagenomic sequencing data have been submitted to the SRA database with BioProject ID PRJNA1138444 and SRA submission ID SUB14608980. Species annotation information at different taxonomic levels (kingdom, phylum, class, order, family, genus, and species) was obtained based on the national center for biotechnology information species classification system. Annotation information was combined with unigene abundance to obtain abundance data at each taxonomic level. At the kingdom level, the vast majority of species were bacteria, among which, Bacteroidetes, Firmicutes, and Proteobacteria were identified as the three most abundant bacterial phyla (Fig. 1). Species diversity analysis revealed no significant differences between the two groups with respect to gut bacterial diversity (i.e., α-diversity or β-diversity).
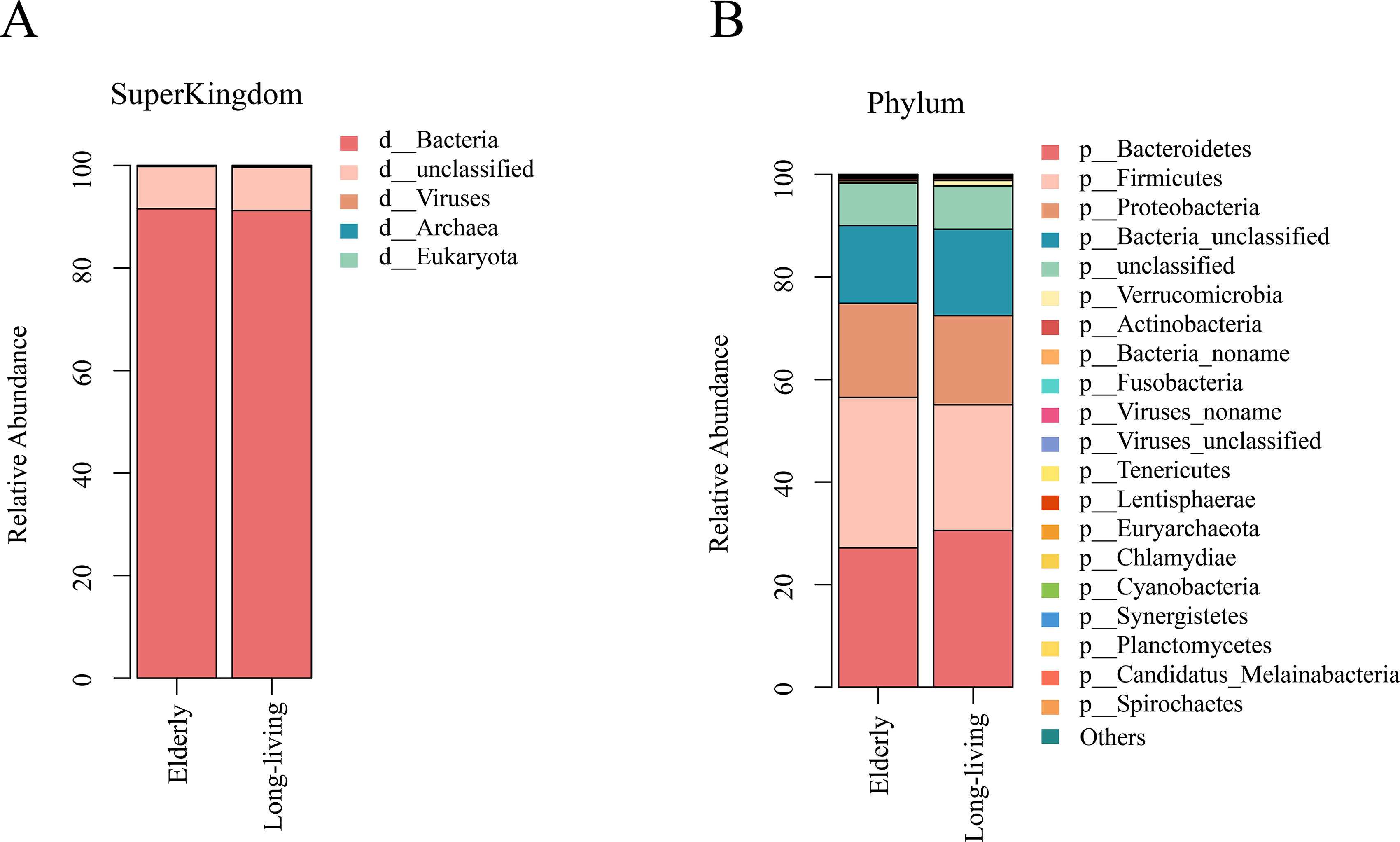
The main composition of the gut microbiota at the kingdom
Analysis of functional differences in the gut microbiota
We detected 63 GO biological processes differing significantly in relative abundance between long-living and elderly individuals (Supplementary Table S1), including an enhancement of the following biological processes in the long-living individuals: autophagy, telomere maintenance using semiconservative replication, melanin metabolism, response to fungi and nematodes, regulation of signal transduction through tumor protein P53 class mediators, secretion of lysosomal enzymes, positive regulation of intracellular cholesterol transport, and activation of mitogen-activated protein kinase (MAPK) activity.
Conversely, long-living individuals were found to have attenuations of the following processes: visual perception, long-chain fatty acid import, cellular response to transforming growth factor beta stimulus, S-adenosyl methionine metabolism, nicotinamide adenine dinucleotide (NADH) oxidation, chronological cell aging, negative regulation of protein kinase B signaling, negative regulation of endothelial cell differentiation, negative regulation of vascular endothelial growth factor signaling pathway, negative regulation of vasculogenesis, and positive regulation of MAPK 14(p38) cascades (Fig. 2).
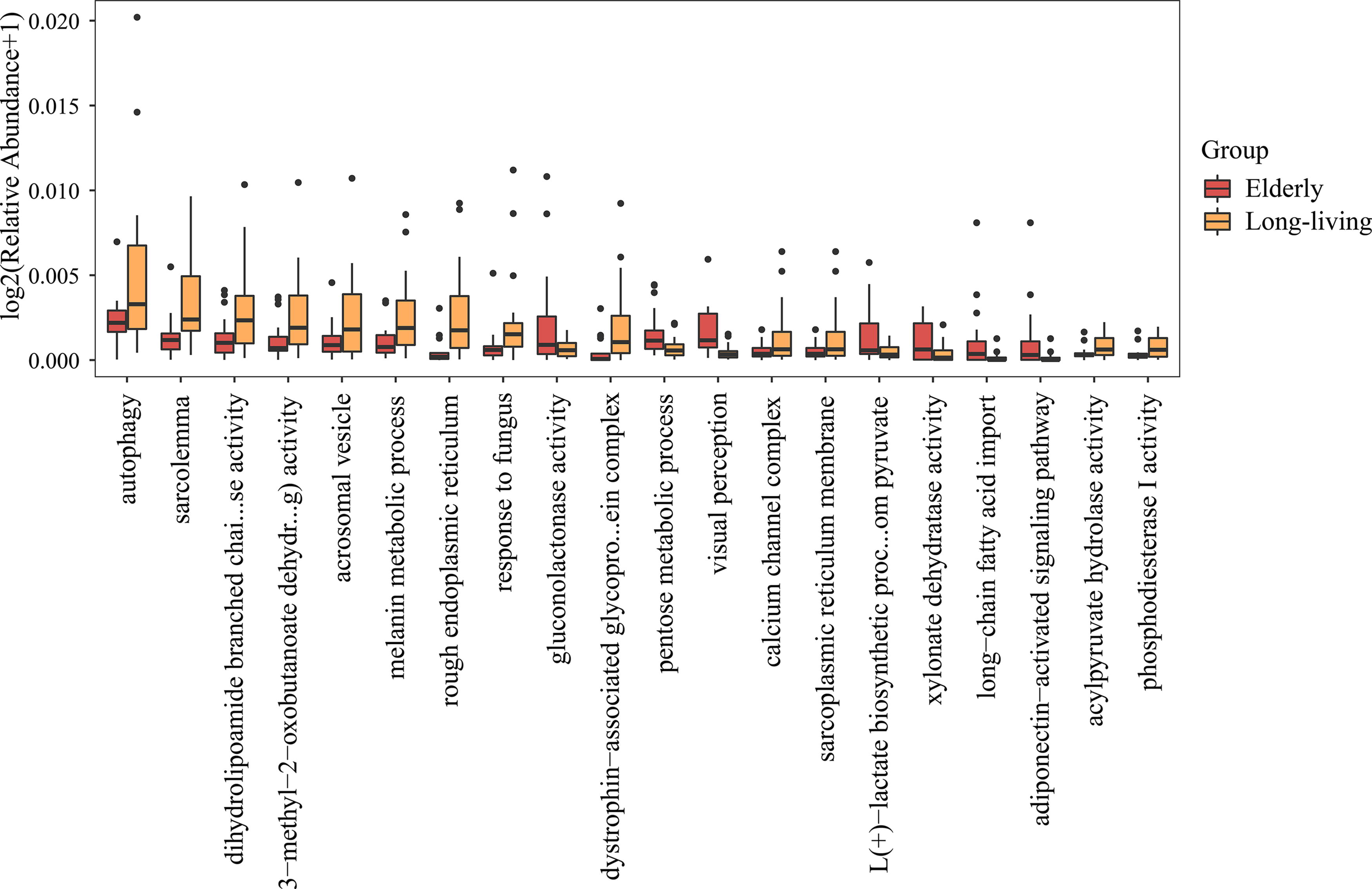
Gene Ontology biological processes differing significantly between the long-living and elderly individuals.
The abundance of each biological process was derived from the abundance of related unigenes, which was also used to count the abundance of gut species. With the help of intermediary (unigenes), we could explore the relationship between the difference in gut species and the functional differences, as well as identify which species caused the functional differences. Supplementary Figure S1 showed the relationship between the abundance difference in gut species and a few biological processes.
In addition, the two groups were found to differ significantly with respect to 10 KEGG pathways, among which there were reductions in the biosynthesis of amino acids; glycine, serine, and threonine metabolism; lysine biosynthesis; aminoacyl-tRNA biosynthesis; glycolysis/gluconeogenesis; protein export; RNA polymerase; and RNA degradation in the long-living individuals, with neomycin, kanamycin, and gentamicin biosynthesis being the only differential pathway enhanced in these individuals.
Differential species/genus analysis
Among the major bacterial phyla, we detected no significant differences between the long-living and elderly groups with respect to the abundances of Firmicutes (24.54 vs. 29.33), Bacteroidetes (30.55 vs. 27.19), or Proteobacteria (17.37 vs. 18.35) (p > 0.05). In contrast, at the genus level, we identified a total of 29 genera differing significantly between the two groups, among which 11 and 18 were up- and downregulated, respectively, in the long-living individuals.
At the species level, the two groups differed significantly with respect to 124 species, with 36 species being upregulated and 88 downregulated (e.g., Bacteroides plebeius and Sutterella wadsworthensis) (Supplementary Table S2). Table 1 listed only differential genera and species with a mean abundance over 0.01. In addition, LEfSe (Linear discriminant analysis Effect Size) analysis revealed significant reductions in the relative abundances of the species Megamonas funiformis, Megamonas_unclassified, and Bacteroides plebeius; genus Megamonas, family Selenomonadaceae; and order Selenomonadales in the long-living individuals.
Bacterial Genera/Species Differing Significantly In Relative Abundance Between Long-Living and Elderly Individuals
Only differential genera and species with a mean abundance over 0.01 are shown.
Classification marker screening
The top 20 species deemed to be of classification importance, based on analysis using a random forest model, are presented in Figure 3A. The x-axis term “MeanDecreaseAccuracy” denotes the degree of reduction in the predictive accuracy of the random forest model when the data for a variable are shuffled, with larger values for variables being taken to be indicative of a greater importance. The relative abundance of these 20 species also differed significantly between the two groups (Fig. 3B), indicating that these species could be used as potential classification markers to distinguish between the long-living and elderly populations.

The top 20 most important classification markers generated by random forest model
Analysis of differences in unigene expression and functional enrichment
In total, we detected 11,590 and 22,363 unigenes that were up- and downregulated, respectively, in the long-living individuals. GO and KEGG enrichment analyses revealed that these differentially expressed unigenes were mainly enriched in biological processes such as phosphorylase signal transduction system, regulation of transcription, DNA template, DNA repair, proteolysis, carbohydrate metabolic process, glycolytic process, fatty acid biosynthetic process, arginine biosynthetic process, and methionine biosynthetic process (Fig. 4A). Notably enriched pathways included biosynthesis of amino acids, microbial metabolism in diverse environments, biosynthesis of secondary metabolites, biosynthesis of cofactors, carbon metabolism, and glycolysis/gluconeogenesis (Fig. 4B).
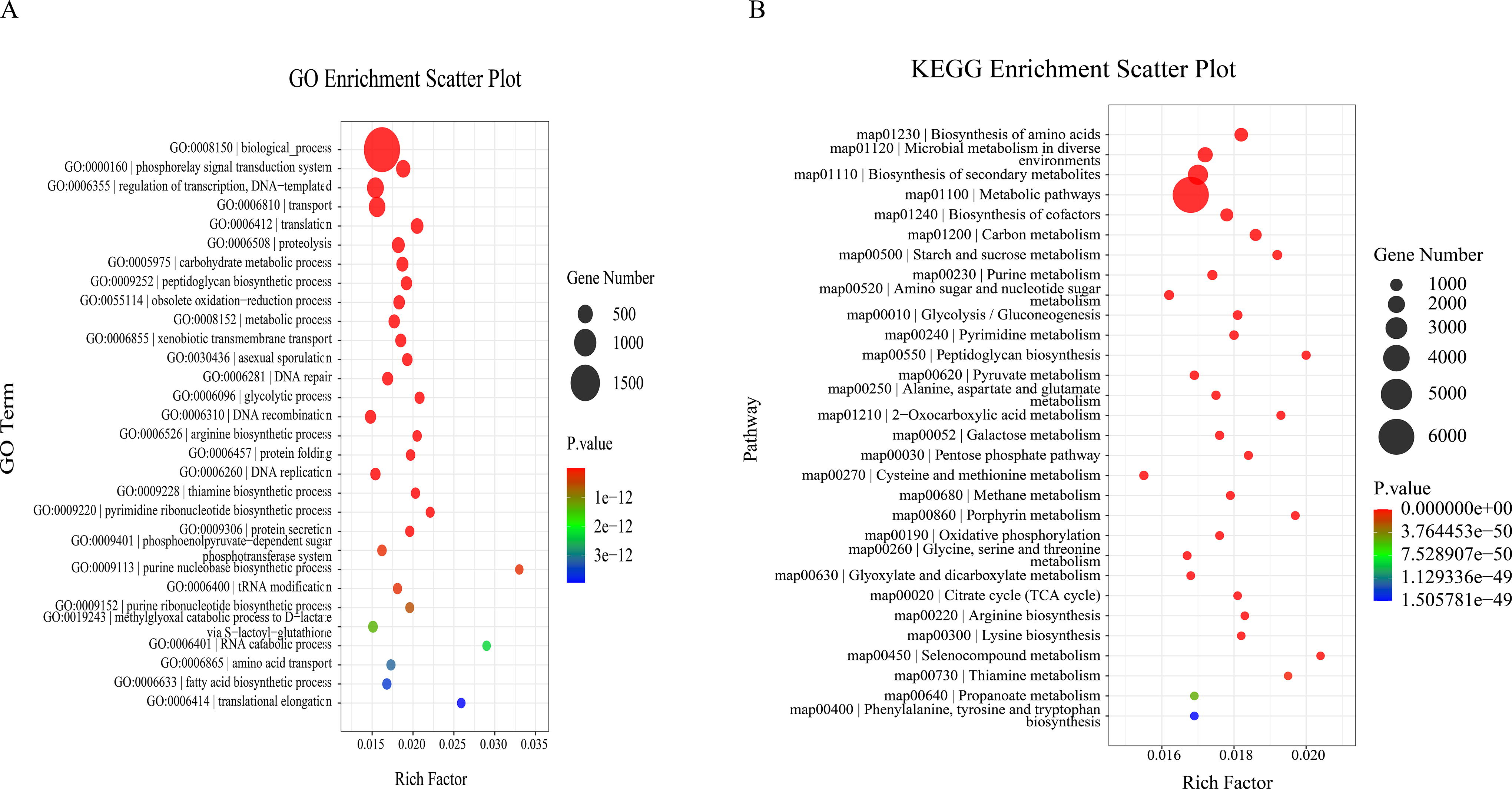
Results of GO
Discussion
With a view toward identifying gut microbiota signatures associated with aging and longevity, as study subjects, we selected adults over the age of 90 and those aged between 65 and 74 years from the inhabitants of Kaihua County, Quzhou City, designated as the “capital of longevity.” To characterize the respective gut microbiota and examine differences between the two groups, we performed metagenomic sequencing. Compared with 16S rRNA sequencing (Ren et al., 2021; Wang et al., 2019; Yu et al., 2015), metagenomic sequencing facilitates in-depth studies at the genetic and functional levels (e.g., GO functional and KEGG pathway analyses) and can be used to characterize microorganisms down to the species or strain level.
Notably, we detected no significant differences between the two subject groups with respect to gut microbial diversity, which tends to contrast with the findings of a number of previous studies that have reported a higher diversity of gut microbiota in long-living individuals (Kong et al., 2016), and could thus provide evidence to indicate that a higher abundance of beneficial bacteria and richer microbial diversity may be important factors contributing to longevity and the maintenance of human health (Kong et al., 2019). We suspect that our inability in the present study to detect significant differences in the latter context between long-living and younger elderly individuals could be associated with the small sample size and the comparatively small geographical area from which these individuals were drawn.
In terms of the composition of the gut microbiota, we detected a significant reduction in the relative abundances of certain genera and species in long-living individuals, including the bacterial genera Sutterella and Megamonas. Species of Sutterella are among the most abundant in the phylum Proteobacteria that play important roles as commensal bacteria in the intestinal tract, being abundant in the duodenum of healthy adults and gradually declining in abundance toward the colon. They have a mild pro-inflammatory capacity in the human gastrointestinal tract. There is some evidence to indicate that by secreting immunoglobulin A(IgA) proteases, and thereby causing a reduction in IgA concentrations in the intestinal mucosa, Sutterella may impair the function of the intestinal antimicrobial immune response (Kaakoush, 2020). However, the findings of other studies have tended to indicate that these bacteria do not induce a substantial inflammatory response. Given the assumed importance of chronic inflammation in the aging process, the roles of Sutterella in aging and longevity warrant further investigation from a pro-inflammatory perspective.
Species of Megamonas, a genus of gram-negative bacteria in the phylum Firmicutes, have been characterized as fermenters of a range of carbohydrates, giving rise to end products such as acetic, propionic, and lactic acids. As core intestinal bacteria, species of Megamonas are closely associated with inflammatory bowel disease, colorectal cancer (Yachida et al., 2019), ankylosing spondylitis (AS), autism spectrum disorder (Zou et al., 2020), and obesity (Duan et al., 2021). In addition, a study comparing the composition of the gut microbiota in frail and control group older adults reported lower levels of Megamonas species in the frail group, and hence, the authors speculated that a reduction in the abundance of these bacteria might be associated with frailty (Xu et al., 2021). Species of Megamonas produce short-chain fatty acids, which, upon entering the systemic circulation through intestinal epithelial cells, can provide energy for these cells, thus contributing to the maintenance of intestinal mucosal barrier integrity, immune system regulation, and a reduction in inflammatory responses and, thereby, lending support to the hypothesis that a reduction in Megamonas might be associated with inflammation and frailty in older adults. The lower abundance of Megamonas in the long-living group assessed in the present study may reflect an intrinsic factor contributing to the aging process and could be researched in the future as a potential target for antiaging intervention.
In addition to differences in gut microbial composition, we analyzed functional differences between the gut microbiota in long-living and elderly individuals. The findings of GO functional analysis revealed that many of the biological processes showing differential enrichment between the two populations are closely associated with aging and longevity, among which, those potentially contributing to longevity, including autophagy (GO:0006914) (Aman et al., 2021) and telomere maintenance through semiconservative replication (GO:0032201) (Bernadotte et al., 2016), were found to be enriched in the long-living individuals. Furthermore, in these individuals, we also identified differences with respect to certain processes characteristic of aging, including diminished visual perception (GO:0007601). There were also an attenuation of the response to lipopolysaccharide (GO:0032496), NADH oxidation (GO:0006116) (Chu and Raju, 2022; Salekeen et al., 2021), and S-adenosyl methionine metabolism (GO:0046500) (Loenen, 2018; Thapa et al., 2023) in long-living group.
Lipopolysaccharide (LPS) is a component of the outer cell wall of gram-negative bacteria that mainly comprises lipids and polysaccharides, high levels of which have been detected in the plasma and feces of old mice. Moreover, the LPS component of fecal lysates has been demonstrated to promote the expression of cyclin dependent kinase inhibitor 2A(P16) and Sam and Hd domain containing deoxynucleoside and induce the activation of nuclear factor kappa-B (NF-κB) in mouse peritoneal macrophages in a TLR4-dependent manner, thereby providing evidence to indicate that intestinal microbial LPS may accelerate inflammatory-associated aging (Kim et al., 2016).
Bacterial infections and an accumulation of LPS are also characteristic features of patients with atherosclerotic lesions, and it has been speculated that LPS may exacerbate the pathogenesis of atherosclerosis by inducing and enhancing the senescence and senescence-associated secretory phenotype (SASP)-related inflammatory properties of specific vascular cells in atherosclerotic lesions (Suzuki et al., 2022).
A SASP has been found to promote ADP-ribosyl cyclase 1(CD38)-dependent catabolism of nicotinamide adenine dinucleotide (NAD+) by immune cells, and reduced levels of NAD+ levels can in turn promote senescence. During senescence, the levels of cellular NAD+ and NAD+/NADH ratios are regulated using a number of different pathways, including NADH oxidation, and the disruption of any of these processes can lead to elevated levels of cytoplasmic NADH, adenosine 5’-monophosphate (AMP)-activated protein kinase activation, and an induction of senescence (Chini et al., 2024; Schiuma et al., 2024; Wiley and Campisi, 2021).
S-adenosyl methionine is a methylation-related methyl donor, the cytoplasmic levels of which determine the levels of histone methylation, particularly that of tri-methylation of lysine 4 on histone H3(H3K4me3), thereby contributing to the maintenance of specific cellular states. Consequently, changes in S-adenosyl methionine levels may influence histone and DNA methylation, thereby influencing intracellular homeostasis and thus potentially human lifespan (Zhu et al., 2022). The aforementioned studies have shown that LPS response (GO:0032496), NADH oxidation (GO:0006116), and S-adenosyl methionine metabolism (GO:0046500) are all associated with senescence.
KEGG pathway analysis revealed significant difference between the two study groups with respect to multiple pathways associated with amino acid metabolism. Amino acids not only serve as the main raw materials for the synthesis of proteins and peptides but also play important roles as signaling molecules involved in the maintenance and regulation of metabolic homeostasis, including aging and longevity. Given the overall changes in amino acid levels that occur with advancing age, it is yet to be established whether any amino acids are specifically associated with aging and longevity.
However, a number of studies have sought to assess the age-related changes in individual amino acids and their roles in aging and longevity. For example, studies that have analyzed serum samples have found that the production of tryptophan, threonine, serine, methionine, and cysteine declines with age, whereas increases have been detected in the levels of tyrosine (Panyard et al., 2022). In terms of the underlying mechanisms, it has been demonstrated that by activating target of rapamycin, certain amino acids, including leucine and arginine, can contribute to shortening lifespans.
Conversely, glycine has been demonstrated to extend the lifespans of rodents, and by undergoing conversion to glycine, serine and threonine could play a similar role. Further studies have indicated that serine supplementation in humans reduces plasma homocysteine levels, which serve as a marker of cardiovascular disease. In this regard, metabolism of serine in the mitochondria leads to the production of nicotinamide adenine dinucleotide phosphate, which protects mitochondria from reactive oxygen species-mediated damage (Canfield and Bradshaw, 2019).
In the present study, we observed significant differences in the amino acid metabolism of gut microbiota in long-living and elderly individuals, particularly with respect to amino acid biosynthesis and the metabolism of glycine, serine, and threonine. However, whether these metabolic changes are simply characteristic features of the aging process or make an active contribution to determining longevity is yet to be discerned. Accordingly, further research is needed to elucidate the effects of gut microbiota on amino acid metabolism and to identify the optimal composition of gut microbiota that is most conducive to promoting human longevity and maintaining metabolic health (Kitada et al., 2019).
Despite new findings, this study has several limitations, notable among which is its cross-sectional design, which makes it difficult to determine whether differences are due to different ages and life stages or gut microbiota characteristics specific to long-living individuals. Moreover, the sample size was small, as was the area of the region from which the study populations were drawn, thereby highlighting the need for further studies based on larger samples and multiregional enrollment.
Conclusions
Although our comparative analysis of the gut microbiota in long-living and elderly individuals revealed no significant differences between these populations with respect to microbial diversity, we did, however, detect significant differences regarding the relative abundances of certain bacterial genera and species, several functional and physiological processes, and metabolic pathways. These findings would tend to indicate that the guts of long-living individuals are characterized by distinct microbial profiles and that the microbiota potentially influence longevity by regulating biological processes such as autophagy, telomere maintenance, and amino acid metabolism. Further studies will, however, be necessary to verify the associations between these differences and longevity, as well as to elucidate the underlying mechanisms of action, the findings of which may contribute to identifying potential targets for antiaging treatments or interventions.
Footnotes
Acknowledgments
Author Contributions
All authors made a significant intellectual contribution, met the International Committee of Medical Journal Editors (ICMJE) criteria for authorship, and agreed to and take public responsibility for the final version of the article.
Author Disclosure Statement
No conflict of interests was reported by the authors.
Funding Information
The work was funded by the Quzhou competitive projects for tackling key scientific and technological problems (2023K200).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.