
Abstract
One Health and planetary health place emphasis on the common molecular mechanisms that connect several complex human diseases as well as human and planetary ecosystem health. For example, not only lung cancer (LC) and gastroesophageal reflux disease (GERD) pose a significant burden on planetary health, but also the coexistence of GERD in patients with LC is often associated with a poor prognosis. This study reports on the genetic overlaps between these two conditions using systems biology-driven bioinformatics and machine learning-based algorithms. A total of nine hub genes including IGHV1-3, COL3A1, ITGA11, COL1A1, MS4A1, SPP1, MMP9, MMP7, and LOC102723407 were found to be significantly altered in both LC and GERD as compared with controls and with pathway analyses suggesting a significant association with the matrix remodeling pathway. The expression of these genes was validated in two additional datasets. Random forest and K-nearest neighbor, two machine learning-based algorithms, achieved accuracies of 89% and 85% for distinguishing LC and GERD, respectively, from controls using these hub genes. Additionally, potential drug targets were identified, with molecular docking confirming the binding affinity of doxycycline to matrix metalloproteinase 7 (binding affinity: −6.8 kcal/mol). The present study is the first of its kind that combines in silico and machine learning algorithms to identify the gene signatures that relate to both LC and GERD and promising drug candidates that warrant further research in relation to therapeutic innovation in LC and GERD. Finally, this study also suggests upstream regulators, including microRNAs and transcription factors, that can inform future mechanistic research on LC and GERD.
Introduction
The emerging intersection of planetary health, One Health, and systems biology not only challenges the binary of human and ecosystem health but also invites thinking beyond the disease silos (Biswas et al. 2023; Dasgupta, 2024a; Dasgupta et al. 2023). Chief among these possibilities is the idea of exploring the pathophysiological mechanisms shared across diseases.
In this overarching context, it is noteworthy that lung cancer (LC) accounts for almost 2 million cases worldwide and 1.8 million deaths annually and is one of the most common cancer subtypes with a high mortality and morbidity rate (Thandra et al., 2021). Small cell lung cancer (SCLC) and nonsmall cell lung cancer (NSCLC) are the two histological subtypes of LC that can potentially benefit from One Health, planetary health, and systems biology research and conceptual frames. Of note, NSCLC makes up around 85% of all cases, whereas SCLC makes up the remaining 15% (Schabath and Cote, 2019). While several risk factors increase the likelihood of a poor outcome and cancer progression, emerging reports interestingly indicate a significant positive correlation between gastroesophageal reflux disease (GERD) and LC prognosis (Amarnath et al., 2022; Ghanem et al., 2007). In a Taiwanese population sample, patients with GERD exhibited a 50% higher risk of developing LC as compared with those without GERD (Amarnath et al., 2022). Another population-based study conducted in Taiwan, which includes 42,555 subjects, also reveals that patients with GERD have a considerably greater prevalence of LC than the controls (hazard ratio = 1.53; Hsu et al., 2016). Vereczkei et al. (2008) reported that the prevalence of GERD in patients with NSLC is significantly higher than in the noncancerous group. Interestingly, a meta-analysis by Putra et al. and a Mendelian randomization study by Li and his group also confirmed a significant positive association between LC and GERD (Li et al., 2023; Putra and Putra, 2021).
Prolonged secretion of stomach acid in patients with GERD has been suggested to lead to changes in the lining and potentially progress to carcinoma (Hsu et al., 2016). Even though GERD is noted as a risk factor for the progression of LC, no studies to date have reported genetic/genomic overlaps between these two conditions. These clinical observations invite further mechanistic research, for example, by unpacking the correlation between LC and GERD in relation to genetic/genomic biomarkers.
Using systems biology-based approaches for interpreting publicly available Gene Expression Omnibus (GEO) datasets offers a promising path for identifying common gene signatures and associated pathways in various disorders with overlapping mechanisms. Identification of hub genes, that is, genes that are tightly connected in a given regulatory network and associated with disease pathogenesis, can also highlight potential targets for therapeutic intervention and thus provide deeper insights into the molecular mechanisms underlying these disorders.
A search of the PubMed in August 2024 indicated a total of 48 studies that highlight differentially expressed genes (DEGs) in LC using bioinformatics analysis in the last 1 year. Several genes including COL1A1, THRα1, and SPP1 were found to be significantly altered in patients with LC as compared with controls (Geng et al., 2021; Matsubara et al., 2023). In the case of GERD, a total of seven bioinformatics-based studies are reported. Zhao et al. (2021) highlighted the role of resolvin D1 in the pathogenesis of GERD and the potential role of this gene in acid-induced DNA damage in esophageal epithelial cells in patients with refractory GERD and a rat model of acid reflux. Despite studies suggesting DEGs in individual diseases, LC versus controls and GERD versus controls, there exists no study to the best of my knowledge at the time of writing of the present report in August 2024 where genetic correlations between these two conditions have been reported.
MicroRNAs (miRNAs) and transcription factors are critical in regulating gene expression and influencing the onset and progression of various diseases (Qin et al., 2020). Identifying specific miRNAs and transcription factors associated with overlapping genes between LC and GERD can unpack the regulatory networks and potential therapeutic targets (Yu et al., 2020). Many reports have also revealed that combining genetic expression data with machine learning-based algorithms can markedly enhance the predictive power of genetic markers (Dasgupta et al., 2023; Sherpa et al., 2024; Vadapalli et al., 2022). Random forest (RF) and K-nearest neighbor (KNN) algorithms are two promising approaches for the selection of important features from high-dimensional genomic data (Joon et al., 2023; Pragya et al., 2023). In RF applications, the input variables are primarily the features derived from gene expression data. These input variables can be quantitative and continuous, representing the expression levels of various genes across the samples. The response variable in this model is categorical, typically indicating the disease status, such as distinguishing between LC and GERD from controls. The algorithm controls these input features to build multiple decision trees and aggregate their predictions to classify the samples accurately. RF is referred to as a multivariate model because it simultaneously considers multiple input features (genes) to make predictions, capturing complex interactions between them.
For the KNN algorithm, the input variables are also the gene expression levels, which are quantitative and continuous. These variables represent the expression profiles of genes across different samples. The response variable is categorical, indicating the classification outcome, such as whether a sample belongs to the LC or GERD or control group. KNN is also considered a multivariate model as it uses the entire set of input features to determine the similarity between samples. By evaluating the proximity of a sample to its nearest neighbors in the feature space, KNN classifies the sample based on the majority class among these neighbors, effectively considering multiple variables in the classification process. Therefore, the present research aims to highlight the regulators as well as the classification predictability of the common hub genes to differentiate LC and GERD from controls.
In addition to machine learning and biological association analysis, the advancements in structural biology have also significantly enriched our ability to determine potential drugs targeting candidate genes associated with complex disorders (Dasgupta, 2024b). Molecular docking, an advancement in structural bioinformatics, provides insights into the binding affinity between a target protein and potential small molecules/drugs, aiding in rational drug design (Dasgupta and Ghosh, 2024; Gundampati et al., 2012). In addition to the binding analysis, the pharmacokinetic characteristics, including absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the identified drug, can further highlight its suitability inside the organism (Ferreira and Andricopulo, 2019). It is, therefore, envisioned that the determination of candidate drugs targeting overlapping genes between LC and GERD, coupled with detailed structural analysis and assessment of pharmacokinetics and bioavailability characteristics, holds veritable promise for rational drug discovery targeting genetic markers that overlap in several diseases.
In the present study, the genetic commonalities between LC and GERD using bioinformatics-based analyses are examined. The hub genes that are associated with both disorders are unpacked, and the expression of these genes was validated in additional datasets. Multivariate models further revealed the importance of the identified genetic predictors for classifying both LC and GERD from controls. The upstream regulators including miRNAs, transcription factors, and pathways associated with these hub genes are also evaluated. Subsequently, the promising drugs that target these common hub genes between LC and GERD are predicted. The pharmacokinetics, toxicity, and bioavailability of the candidate drug that targets the hub gene are determined. Finally, the binding affinity of this drug with the targeted protein is reported using molecular docking analysis.
Materials and Methods
Publicly available datasets were used in the present study, and informed consent and research ethics board approval were not required. The study was conducted under the overall research ethics oversight of the author’s institution.
Identification of the common DEGs
The publicly available database NCBI-GEO (http://www.ncbi.nlm.nih.gov/geo/) was searched using the keywords “lung cancer” and “gastroesophageal reflux disease” (accessed June 4th, 2024). The datasets were shortlisted based on two criteria: (1) availability of the full dataset and (2) datasets belonging to “Homo sapiens.” Through extensive research, two datasets, GSE268175 and GSE226303, were selected for patients with LC and GERD.
The GSE268175 dataset contains RNA-sequencing data from 34 early-stage NSCLC samples, as well as adjacent noncancerous tissues. The patients underwent surgical procedures at three different clinics (Dushu Lake Hospital Affiliated to Soochow University, the First Affiliated Hospital of Soochow University, and Sichuan Cancer Hospital). The patient population is fairly distributed between those younger than 65 years (38%) and those 65 years or older (62%). There is a balanced gender distribution, with 38% of the patients being female and 62% male. In addition, the body mass index (BMI) is equally split, with 50% of patients having a BMI < 24 and 50% having a BMI of 24 or greater. Tissue samples ranging from 100 mg to 500 mg were obtained from the recruited and used to generate RNA-sequencing libraries from these subjects. Subsequently, paired-end sequencing of the libraries was performed on the Illumina HiSeq-PE150 instrument. The clinical characteristics of the recruited subjects are provided in Supplementary Table S1 (Mao et al., 2024).
The dataset GSE226303 comprises individuals experiencing heartburn symptoms, who underwent phenotypic characterization through endoscopy and objective reflux studies. The subjects were recruited at the Royal London Hospital (Queen Mary University of London). The average age of the recruited subjects varies across different phenotypes, with the highest mean age observed in patients with Barrett’s esophagus at 54 years and the lowest in healthy controls at 27 years. RNA was extracted from esophageal mucosal biopsies samples of 37 patients with GERD and 8 controls. Following the extraction, RNA samples were sequenced using the Illumina NextSeq 500 platform, and the data were analyzed using Partek Flow and xCell software. The baseline characteristics of the recruited subjects are provided in Supplementary Table S2 (Ustaoglu et al., 2023). It is ensured that both datasets used in the present study are RNA sequencing datasets to maintain consistency in gene expression data, enabling accurate and reliable comparisons between the genetic profiles of LC and GERD. The study design workflow is depicted in Figure 1.
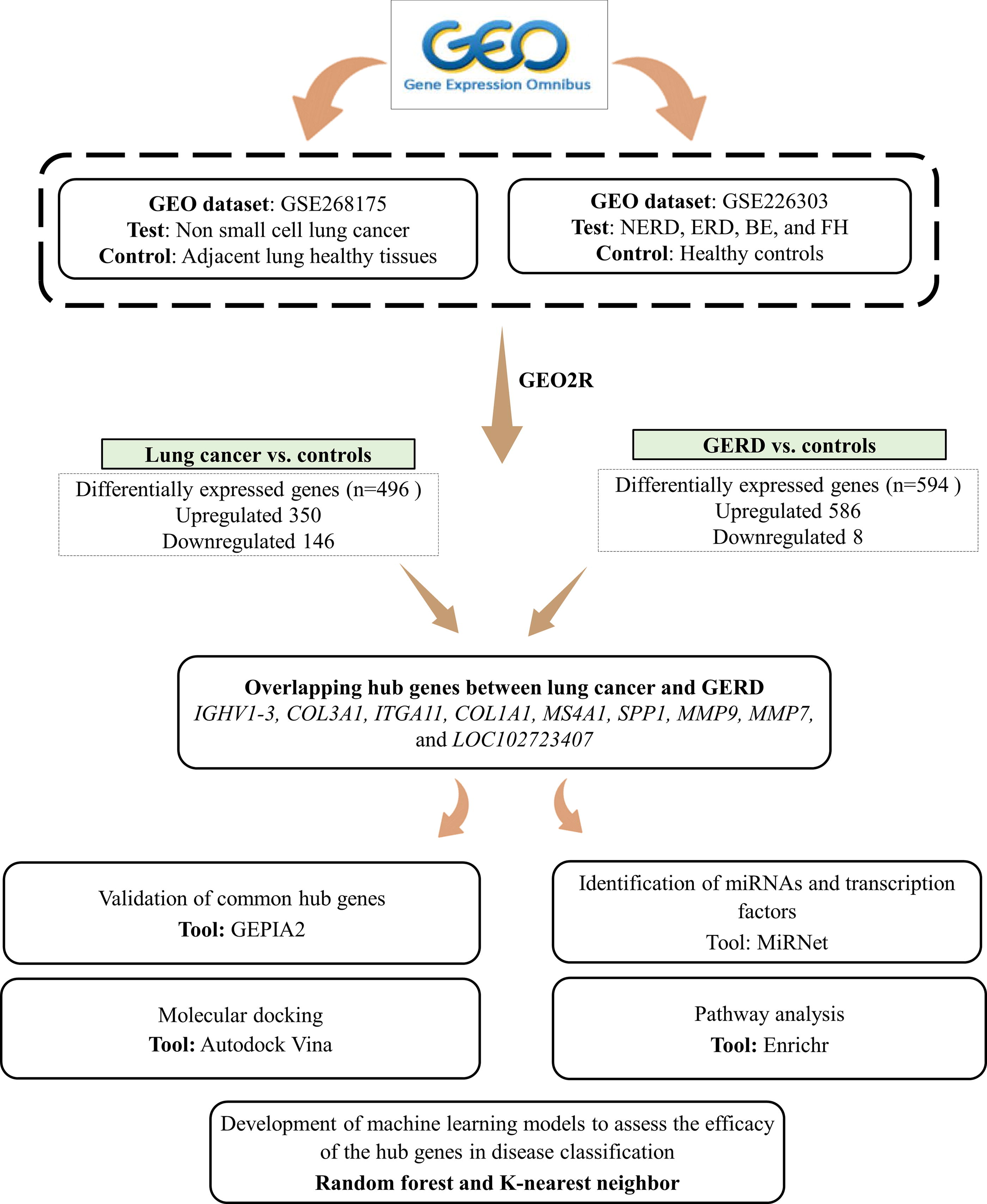
Workflow of the study design. Two Gene Expression Omnibus (GEO) databases were selected to identify the overlapping differentially expressed genes between lung cancer and gastroesophageal reflux disease. NERD, nonerosive reflux disease; ERD, erosive reflux disease; FH, functional heartburn; BE, Barrett’s esophagus.
Screening of common DEGs
The genome data procured from the NCI-GEO tool were analyzed using the GEO2R package, which utilizes limma and GEOquery algorithms for comparative gene expression analysis. In accordance with the definitions implemented in November 2020, groups for comparison are defined in the Samples panel during GEO2R analysis. The order in which groups are defined affects the log fold changes, with the test group (LC/GERD) typically defined first to ensure positive values for upregulated genes. For DEG identification, the tool incorporates false discovery rate (FDR) adjustment to address multiple data testing. In the present study, the Benjamini–Hochberg procedure is used for the FDR adjustment (adjusted p value <0.01). Next, the genes with ∣log2(fold change) ∣ > 2 and p value < 0.05 were considered as the DEGs in both patients with LC and GERD as compared with controls. The overlapping DEGs between these two diseases were visualized by plotting a Venn diagram.
Hub genes identification
It is reported that hub genes are those that are highly interconnected with each other in the biological network and play a crucial role in gene regulation and biological processes (Kang et al., 2022). A novel algorithm of Cytoscape termed cytoHubba offers various topological algorithms to screen the hub genes. Following previous studies, three topological calculation methods: maximal clique centrality (MCC), maximum neighborhood component (MNC), and density of maximum neighborhood component (DMNC) were used to determine the hub genes (Cheng et al., 2023; Choudhury et al., 2024). MCC measures the centrality of genes within densely connected subnetworks, MNC evaluates the influence of genes based on their immediate neighborhood within the network, and DMNC assesses the density and centrality of genes’ neighborhoods in the network. The genes overlapping in all three algorithms were termed the hub genes for LC and GERD cases (Shannon et al., 2003).
Gene and protein–protein interaction network
To construct the gene network of the common hub genes, a user-friendly web interface GeneMANIA was used (http://www.genemania.org). The coexpression and colocalization among the identified hub genes can be explored from this network (Franz et al., 2018). Besides the gene network, the protein–protein interaction network was also developed. Another web tool, STRING (https://string-db.org/), which integrates diverse sources of biological data to predict functional associations among proteins, was used for this purpose (Dasgupta and Ghosh, 2024). This network provides insights into the complex interplay between proteins, offering a comprehensive view of their interactions and potential functional relationships (Szklarczyk et al., 2017).
Hub gene–miRNA-transcription factor network and pathway analysis
The MiRNet tool (https://www.mirnet.ca/), an integrated platform connecting miRNAs and transcription factors with genes, was utilized to predict the miRNAs–DEGs and transcription factors–DEGs interaction network. This integrated platform connects miRNAs with genes and transcription factors, helping to construct and analyze interaction networks. The interaction data of miRNet were collected from three databases, including miRTarBase v7.0, TarBase v7.0, and miRecords, which are catalogs that predict genes and miRNAs as well as genes with transcription factor association (Chang et al., 2020). A cutoff value of degrees >5 was applied to detect the associated miRNAs. The transcription factors exhibiting degrees >1 with hub genes were selected. Based on the degree and betweenness centralities, the most connected miRNAs and transcription factors with the common hub genes were identified.
Pathway analysis offers insights into the biological pathways that are enriched with hub genes, providing a comprehensive understanding of the underlying molecular mechanisms in LC and GERD. Enrichr (https://maayanlab.cloud/Enrichr/) was utilized to investigate the pathways associated with the nine common hub genes (Kuleshov et al., 2016). The pathway that is most significantly associated with these common hub genes is indicated.
Predictive model using machine learning classifier
RF classifier (number of trees = 50), a dimensional reduction machine learning approach, was established to map the scores of important genetic features in the classification of LC and GERD from controls. The accuracy of the prediction model to differentiate the disease groups from controls based on the hub genes was assessed using the Scikit-learn package in Python 3.8 (https://www.scikit-learn.org/stable; Ghosh et al., 2021). The accuracy of the predicted model was also investigated using another supervised machine learning approach, KNN.
Identification of potential drugs targeting overlapping proteins
To explore potential drugs targeting the common dysregulated genes, DGIdb (Drug Gene Interaction Database) was utilized. DGIdb, a drug–gene interaction database (https://www.dgidb.org/search_interactions), comprises data related to human drugs, “druggable genes,” and “drug–gene interactions” from various sources and currently contains more than 14,144 drug–gene interactions between 2611 human genes and 6307 drugs (Wagner et al., 2016). The hub gene names were provided in the interface, and the list of the potential drugs that target these genes was generated.
ADMET analysis
The pharmacokinetic properties of the potential drug were determined using ADMET analysis. SWISSADME, an online server, was used to determine the ADMET properties of the potential drugs with the probable best matches (http://www.swissadme.ch/) (Waterhouse et al., 2018). The PubChem web tool was used to obtain the simplified molecular-input line-entry system (SMILES) format of the identified ligands/drugs (https://pubchem.ncbi.nlm.nih.gov/).
Toxicity and bioavailability prediction
The toxicity level of the predicted drugs was investigated using the Protox-II web tool (https://tox-new.charite.de/protoxII/). The tool contains computer-based models trained on real data (in vitro/in vivo) to predict the toxic potential of the existing and virtual compounds. The acute toxicity class and different endpoints are calculated for an input compound based on the chemical similarities to toxic compounds and trained models (Banerjee et al., 2018). The result of toxicity analysis shows the predicted median lethal dose 50 (LD50) in mg/kg weight, toxicity class, and prediction accuracy of the drug compounds investigated.
Molecular docking
Preparation of protein and drug
The preparation of the target protein was performed according to the study by Tallei et al. (2020). In the present study, the crystal structure of the target protein was retrieved from the RCSB Protein Databank (http://www.rcsb.org/pdb/). Next, three-dimensional structures of the identified drug were obtained from the PubChem server (https://pubchem.ncbi.nlm.nih.gov/compound). The structure of the candidate drug was procured in simple data format.
Docking of drugs into protein
AutoDock Vina (UCSF-Chimera©version1.13) software was used to dock the drug into the target protein. First, the ligand compounds were uploaded in this tool and prepared for docking analysis by adding hydrogen bonds as well as Gasteiger charges. Similarly, the Protein Data Bank (PDB) file of the receptor was uploaded, and the water molecules were removed. The hydrogen bonds and Gasteiger charges were added to the receptor. The docked protein was consequently saved as a pdbqt file and then subjected to the docking process. A total of 10 configurations with the best docking poses for each protein–ligand complex were generated. The compounds with the lowest binding energy (kcal/mol) and minimum root mean square deviation (RMSD) were considered the most suitable docking pose since they represent the most significant interaction (Trott and Olson, 2010).
Results
Overlapping DEGs identification in LC and GERD
Following the processing of the raw data using the GEO2R package, a total of 496 DEGs including 350 upregulated and 146 downregulated genes as compared with controls were identified in patients with LC. In the case of GERD, 586 genes are upregulated and 8 are downregulated genes as compared with healthy subjects. The Venn diagram constructed with the altered genes suggested that a total of 72 genes were significantly upregulated in both LC and GERD as compared with controls (Supplementary Fig. S1).
Selection of nine hub genes
The hub genes were selected by overlapping the genes obtained from three topological analysis methods, that is, MCC, MNC, and DMNC. A total of nine hub genes including IGHV1-3, COL3A1, ITGA11, COL1A1, MS4A1, SPP1, MMP9, MMP7, and LOC102723407 were found to be significantly upregulated in both LC and GERD as compared with controls.
Network and enriched pathway analysis
GeneMANIA indicates that the interaction between the overlapping hub genes has 12.87% physical interaction and 28.42% coexpression (Supplementary Fig. S2). Further, the STRING online database generated the PPI network with 9 nodes and 15 edges, exhibiting a significant enrichment (p value: 4.84e-10) (Supplementary Fig. S2). In addition to the biological networks, the Enrichr tool indicated that the extracellular matrix remodeling pathway is significantly enriched in both LC and GERD (Supplementary Fig. S2; p < 0.05).
Correlation analysis between the common hub genes
The heat map in Figure 2 illustrates the correlation between hub genes of LC and GERD, with correlation values ranging from 0.2 to 0.9. In this visualization, darker red boxes signify a strong association between the gene expression profiles, indicating a high positive correlation. The most intense correlations (darker reds) were observed between COL3A1-COL1A1, COL1A1-ITGA11, and COL3A1-ITGA11.
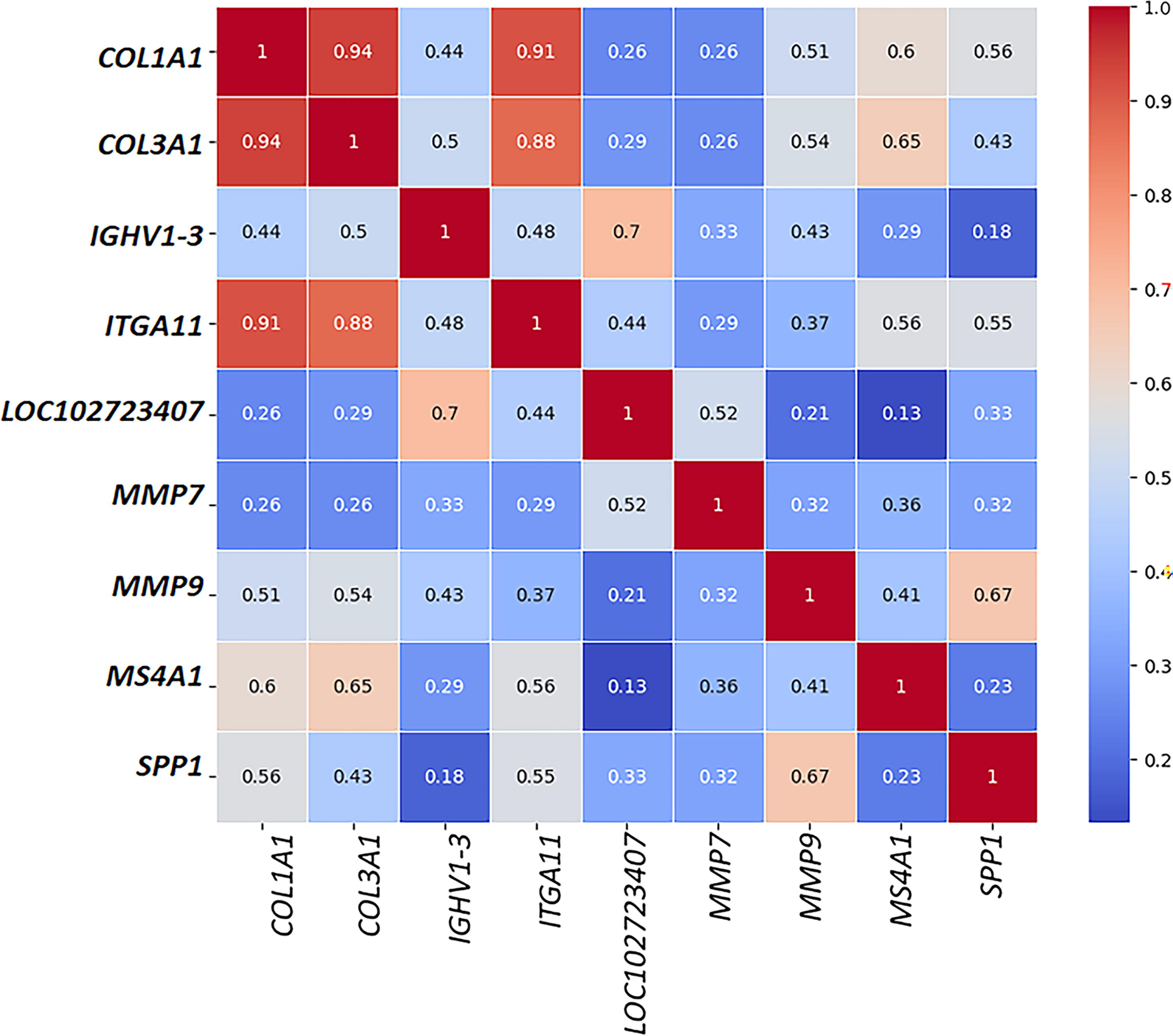
Correlation between the hub genes altered in both lung cancer and gastroesophageal reflux disease.
MiRNA and transcription factors associated with the hub genes
The miRNet interaction network indicated that hsa-miR-126-3p, hsa-miR-34a-5p, hsa-miR-145-5p, and hsa-miR-16-5p are significantly associated with the common hub genes. The transcription factors–hub gene interaction network suggested that three transcription factors including specificity protein 1 (SP1), Jun proto-oncogene, AP-1 transcription factor subunit (JUN), signal transducer and activator of transcription 3 (STAT3), and CCAAT enhancer binding protein alpha (CEBPA) are associated with nine hub genes common between LC and GERD. Supplementary Table S3 demonstrates the miRNAs and transcription factors as well as their corresponding degrees in terms of their association with the hub genes.
Machine learning-based classification models
The RF model demonstrated a high accuracy (accuracy = 0.89) in differentiating the patients with LC from controls using the overlapping hub genes, indicating its robustness and reliability in disease classification (Fig. 3A). For GERD, the RF model exhibited 85% accuracy in disease classification (Fig. 3D).
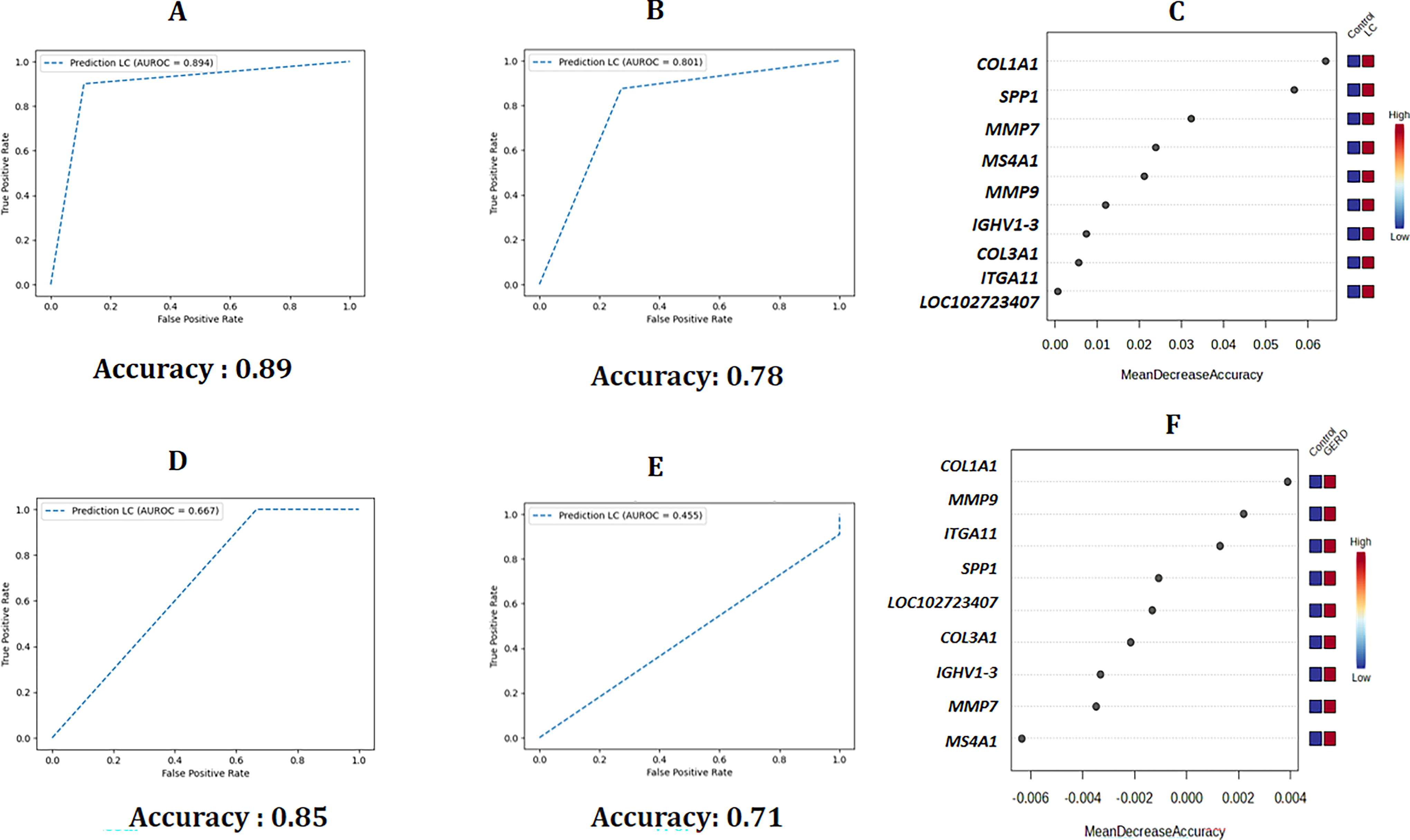
Development of prediction models for classification of lung cancer (LC) and gastroesophageal reflux disease (GERD) from controls.
Furthermore, the KNN algorithm also demonstrated an accuracy of 0.78 for the same classification task, highlighting its strong performance and suitability in the classification of LC from controls (Fig. 3B). It was found that the machine learning model showed an accuracy of 0.71 in differentiating GERD from controls (Fig. 3E). Combing these findings, it was evidenced that these hub genes are effective for distinguishing patients with LC and GERD from controls, with the RF model offering a marginal advantage in discriminatory power. It is also important to note that a feature ranking method was used to identify the most important genes for the classification of disease groups. The genetic feature ranking plots for both LC and GERD are also demonstrated in Figure 3C and 3F. It was interesting to note that COL1A1 exhibited the highest importance in classifying both diseases from controls.
Validation of hub genes
The expression of the hub genes of LC was validated using the Gene Expression Profiling Interactive Analysis tool. For GERD, the GSE246323 dataset was used for the validation purpose (Kleuskens et al., 2024). Both databases include gene expression data obtained from Illumina RNA sequencing platforms. The expression of these genes exhibited a similar type of trend as the discovery cohort in both cases. Supplementary Figure S3 shows the expression of the hub genes in the validation cohort. The boxplot displays median values, quartiles, and potential outliers, facilitating the comparison of gene expression between tumor and normal tissues. Statistical significance is typically assessed using t tests or analysis of variance, highlighting genes with notable expression changes. This observation strengthens our initial findings, and it could be envisioned that targeting this gene with the promising drug can provide a new direction in the treatment strategy of patients with LC associated with GERD.
Identification of potential drugs
The DGIdb webtool was used to identify the drugs having interactions with the nine common hub genes between LC and GERD. It was found that doxycycline has the highest interaction score with one of the hub genes, MMP7. The drug exhibited an interaction score of 1.87 with this gene.
ADMET analysis and toxicity prediction
In the present study, the pharmacokinetic and physiochemical properties of the most promising drug, that is, doxycycline, were performed. In Supplementary Figure S4, the pink zone shows the physicochemical space for oral bioavailability, and the red line defines oral bioavailability properties of the drug. Furthermore, toxicity analysis by Protox II demonstrated that doxycycline belongs to toxicity class-4 with LD50 values of 2240 mg/kg (Supplementary Fig. S4).
Molecular docking
Molecular docking analysis was performed to highlight the binding affinity between matrix metalloproteinase 7 (MMP7) and doxycycline. The RSCB-PDB (https://www.rcsb.org/) database was used to obtain the PDB structure of MMP7 (PDB ID: 2Y6D). The PubChem tool was used to retrieve the three-dimensional structure of the promising drug (doxycycline). Next, utilizing the AutoDock Vina tool, both receptor and ligand were prepared for docking by adding hydrogen and Gasteiger charge. The molecular docking analysis was performed inside a grid box, which is a specialized tool used for predicting the preferred binding position of a ligand to a protein. For the present study, the grid size was set to 103 × 64 × 70 (x, y, z) points with center dimensions (x = 4.77, y = 10.82, z = 8.00 Å), respectively. The specified grid size of 103 × 64 × 70 points in the x, y, and z dimensions, respectively, represents the number of discrete points along each axis within this space. The center dimensions (x = 4.77, y = 10.82, z = 8.00 Å) indicate the central coordinates of this grid precisely in 4.77 Å along the x-axis, 10.82 Å along the y-axis, and 8.00 units along the z-axis from the origin. This setup ensures that the docking algorithm focuses its search for potential binding modes within a defined and appropriately sized region surrounding the protein target.
In the docking procedure, both the ligands and proteins are considered rigid. After the completion of the docking process, 10 configuration files with the 10 best docking poses for each protein–ligand complex were generated. The compounds with the lowest binding energy (kcal/mol) and minimum RMSD were taken as the most suitable docking poses. The binding affinity between MMP7 and doxycycline was −6.8 kcal/mol, indicating a strong interaction between the receptor and the drug (Supplementary Fig. S4). The scores of each docking position are provided in Supplementary Table S4.
Discussion
The coexistence of GERD in patients with LC has been noted in the literature, wherein this comorbidity is significantly associated with a poor prognosis of LC (Amarnath et al., 2022; Ghanem et al., 2007). While two population-based studies indicated a significant association between GERD and the development of LC (Amarnath et al., 2022; Hsu et al., 2016), one meta-analysis and a Mendelian randomization study are also in line with this observation (Li et al., 2023; Putra and Putra, 2021). In contrast, there is a paucity of mechanistic studies and, in particular, of research that focuses on the common genetic overlaps between these two conditions. This study combines systems biology and machine learning-based algorithms to investigate the correlation between LC and GERD, identifying overlapping hub genes and common pathways. In addition to these common hub genes, this study also suggests upstream regulators, including miRNAs and transcription factors, and identifies potential drugs targeting these hub genes through detailed structural analysis. Furthermore, advanced molecular docking algorithms were employed to investigate the binding affinity of the potential drug with the common hub gene.
A total of nine hub genes including IGHV1-3, COL3A1, ITGA11, COL1A1, MS4A1, SPP1, MMP9, MMP7, and LOC102723407 were found to be significantly altered in both LC and GERD as compared with controls. The immunoglobulin heavy chain (IGHV) gene is reported to be associated with metastasis in cancer (Balla et al., 2024). Studies have indicated an association between COL3A1 and COL1A1 with GERD pathogenesis as they are significantly associated with the inflammation of the gastric wall (Chen et al., 2023). In the case of LC, both of these collagens promote tumor metastasis and are reported as prognostic biomarkers (Hou et al., 2021). A recent study by Yang et al. (2024) reported the role of ITGA11 in gastric disorders, as it correlated with increased CD8 + T cell, macrophage, dendritic cell, and NK cell infiltration. ITGA11 is also related to regulating the tumor immune microenvironment and promotes tumor progression by participating in coordinating antigen presentation, cellular immunity, and humoral immunity (Yang et al., 2024).
The role of SPP1 is documented in LC as it actively infiltrates the alveolar macrophages in the tumor environment (Matsubara et al., 2023). Matrix remodeling is a well-known hallmark of the pathogenesis of both LC and GERD, which makes the association between MMP7 and MMP9 with LC particularly plausible. In the case of GERD, MMP7 has been suggested as a potential diagnostic signature, as its expression is directly proportional to the exposure of acidic content secretion and subsequent matrix remodeling (Im et al., 2021). A hypothetical pathway demonstrating the correlation between these genes and the pathogenesis of LC and GERD is shown in Figure 4.

Functional roles of identified hub genes in lung cancer and gastroesophageal reflux disease (GERD). It demonstrates the key processes such as metastasis, where cancer cells spread through the bloodstream, and extracellular matrix remodeling involves COL3A1 and COL1A1. The infiltration and proliferation of T cells and macrophages, linked to ITGA11 and SPP1. Matrix remodeling, crucial for extracellular matrix changes, involves the genes MMP7 and MMP9.
MiRNAs including has-miR-126-3hashsa-miR-34hasp, hsa-miR-145-5p, and hsa-miR-16-5p are found to be significantly correlated with the hub genes associated with LC and GERD. Furthermore, the transcription factor–hub gene network revealed that JUN, STAT3, SP1, and CEBPA are involved in the pathogenesis of both LC and GERD. It is reported that hsa-miR-126, encoded by epidermal growth factor-like domain-containing gene 7, is an endothelial-enriched miRNA and tumor suppressor, playing a major role in the development and metastasis of LC (Chen et al., 2021).
Similarly, hsa-miR-34a-5p is known as a therapeutic target and clinical signature of cancer progression, especially in preventing matrix protein expression to reduce cancer cell mortality (Wen et al., 2022). The altered expression of miR-16-5p is also reported in several malignancies and gastric disorders, highlighting its potential role in inflammatory and apoptotic pathways (Wang et al., 2021). The role of the transcription factors JUN, STAT3, SP1, and CEBPA in LC and GERD is also well established. JUN is a part of the activator protein-1 (AP-1) transcription factor family and is involved in abnormal cell growth and proliferation in cancer cells (Brennan et al., 2020). Therefore, in the case of LC, the overexpression of this transcription factor is significantly correlated with the progression of the tumor. Likewise, STAT3 influences the growth and proliferation of carcinoma cells and apoptotic pathways. Aberrant activation of STAT3 is frequently observed in LC, contributing to oncogenesis and resistance to apoptosis (Xie et al., 2021). The association of SP1 and CEBPA is also reported in LC as it is associated with altered cell differentiation and tumorigenesis (Hsu et al., 2012; Taube et al., 2022). These reports open up a new window into the association between the identified miRNAs and transcription factors with the pathogenesis of LC and GERD.
Notwithstanding their roles in the pathogenesis of LC and GERD, these miRNAs and transcription factors also influence the progression of both disorders. Hsa-miR-126-3p enhances tumor growth through angiogenesis, hsa-miR-34a-5p and hsa-miR-145-5p control cell proliferation and survival, and hsa-miR-16-5p modulates cell cycle and apoptosis, contributing to tumor proliferation and progression of LC (Chen et al., 2021). In the case of GERD, these miRNAs are reported to regulate the esophageal barrier integrity and affect cellular stress and inflammation, thereby playing crucial roles in disease progression (Wu et al., 2020).
Transcription factors including JUN, STAT3, SP1, and CEBPA impact oncogene expression, proliferation, and formation of tumor microenvironment (Brennan et al., 2020). In GERD, the identified transcription factors are reported to drive chronic inflammation and tissue remodeling, exacerbating GERD symptoms (Salas et al., 2020). A hypothetical pathway demonstrating the roles of miRNAs and transcription factors in LC and GERD is shown in Figure 5.

Roles of specific microRNAs (miRNAs) and transcription factors in the pathogenesis of lung cancer (LC) and gastroesophageal reflux disease (GERD). Hsa-miR-126-3p, hsa-miR-34a-5p, hsa-miR-145-5p, and hsa-miR-16-5p are involved in various cellular processes such as cell proliferation, apoptosis, and metastasis. The right section depicts the role of transcription factors, including SP1, JUN, STAT3, and CEBPA, in regulating gene expression and influencing the progression of both LC and GERD.
In the present study, doxycycline exhibited maximum interaction with one of the hub proteins, MMP7. Because MMP7 plays a crucial role in angiogenesis and tumor progression, doxycycline might decline the protumorigenic activities, potentially limiting cancer growth and metastasis. In the context of GERD, the role of MMP7 in matrix remodeling is also significant, as it contributes to the inflammatory responses (Manicone and McGuire, 2008). Hence, doxycycline could reduce the symptoms and progression of both LC and GERD by reducing matrix remodeling and subsequent inflammation.
The present study has several limitations. First, the biological variability present in the samples obtained from LC and GERD may result in disparity in disease-specific fingerprint determination. Also, the quality and consistency of RNA samples obtained from different populations may also vary. Second, the samples used in the present study did not originate from the same population and were procured from two different NCBI-GEO datasets. It is envisioned that future experimental studies with the same patient population cohort will help further ascertain this present observation. Third, the dataset for patients with GERD (GSE226303) includes 37 patients with GERD and 8 controls, whereas the dataset for LC (GSE268175) consists of RNA-sequencing data from 34 early-stage NSCLC samples and adjacent noncancerous tissues. The smaller sample size for LC compared with the GERD dataset is acknowledged, and it is recognized that increasing the number of samples would provide a more robust analysis. Fourth, reversing the datasets used in the present study for analysis and validation showed some impacts on the observations, which is likely due to variations in demographic characteristics, data quality, sample size, and experimental conditions. Finally, the present study reflects bioinformatics and machine learning-based findings. It is important to conduct future experimental studies in several world regions and populations to validate the expression of candidate genes and translate the current findings.
Conclusions
The present study offers novel mechanistic insights into the cooccurrence of GERD in patients with LC. A total of nine hub genes including IGHV1-3, COL3A1, ITGA11, COL1A1, MS4A1, SPP1, MMP9, MMP7, and LOC102723407 were found to be significantly altered in both LC and GERD as compared with controls. The pathway analysis further suggested a significant association between the matrix remodeling pathway and both diseases. MiRNAs including hsa-miR-126-3p, hsa-miR-34a-5p, hsa-miR-145-5p, and hsa-miR-16-5p and transcription factors (JUN, STAT3, and CEBPA) were found to be associated with nine common hub genes between LC and GERD. Interestingly, the machine learning-based models achieved accuracies of 89% and 85% for distinguishing LC and GERD, respectively, from controls using these specific genes. Furthermore, potential drug targets were identified, with molecular docking confirming the binding affinity of doxycycline to MMP7 (binding affinity: −6.8 kcal/mol). Overall, the present study is the first of its kind that combines in silico and machine learning algorithms to identify the gene signatures that relate to both LC and GERD, and promising drug candidates that warrant further research in relation to therapeutic innovation in LC and GERD. Finally, this study also suggests upstream regulators, including miRNAs and transcription factors, that can inform future mechanistic research on LC and GERD.
Footnotes
Acknowledgment
The author is thankful for the permission to use the freely available figure-making tool BioRender.com; GEPIA and NCBI-GEO tool for the gene expression data.
Authors’ Contribution
S.D.: conceptualization, formal analysis, investigation, visualization, and writing–review/editing.
Author Disclosure Statement
The author declares she has no conflicting financial interests.
Funding Information
No funding was received for the present study.
Abbreviation Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.