
Abstract
Lung adenocarcinoma (LUAD) is one of the leading global health challenges wherein novel therapeutic targets are much needed. In this systems bioinformatics study, we report that disruption of the long noncoding RNA (lncRNA) SFTA1P-centered network, respiratory gaseous exchange and surfactant-associated Biological Network (rgsBNet), is consistent with impairing surfactant homeostasis and respiratory function, and thus warrants attention for future drug discovery and development. We analyzed data from The Cancer Genome Atlas LUAD cohort to identify differentially expressed mRNAs, lncRNAs, and microRNAs (miRNAs), followed by correlational analysis to examine the coexpression network of lncRNA SFTA1P and its potential role in LUAD pathogenesis. We observed the downregulation of lncRNA SFTA1P and its coexpressed network in LUAD. Intriguingly, this network appears to be associated with disrupting surfactant homeostasis and perturbing respiratory function, suggesting a potential role in LUAD progression. Additionally, we identified key transcription factors that correlate with the expression of genes crucial for respiratory gaseous exchange and surfactant homeostasis. The attendant regulatory mechanisms suggested that SFTA1P may act as a “sponge” for certain miRNAs, sequestering them away from their mRNA targets. In conclusion, this work uncovers novel insights into the molecular mechanisms governing surfactant homeostasis in LUAD and offers a possible avenue for therapeutic interventions aimed at ameliorating lung function and improving disease management. The downregulation of lncRNA SFTA1P and its coexpressed network highlights their potential as regulators of lung function and opens doors for further investigation into their role in LUAD progression and as potential therapeutic targets.
Introduction
Non-small cell lung cancer (NSCLC) is a frequently diagnosed subtype of lung cancer with a poor prognosis. It constitutes approximately 85% of all lung cancers, whereas small-cell lung cancer comprises nearly 15% (Zappa and Mousa, 2016). Lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), and lung large cell carcinoma are the three primary histological forms of NSCLC arising from different lung tissues. According to the 2020 Global Cancer Observatory (GLOBOCAN) report, 19.3 million new cancer cases and 10 million cancer-related deaths were reported worldwide (Ferlay et al., 2021; Sung et al., 2021). Lung cancer ranked the second highest in cancer diagnoses (11.4%), and it is the primary cause of cancer mortality, accounting for 18% of cancer-related deaths (Ferlay et al., 2021; Sung et al., 2021). Despite considerable advancements in early diagnosis and therapeutic strategies, including targeted chemotherapy and immunotherapy, the 5-year survival rate for NSCLC remains low (Rodak et al., 2021). Additionally, as NSCLC advances to later stages (Stage 3 and Stage 4), survival rates significantly decrease. The estimated 5-year overall survival rates range from 13% to 27% for early-stage diagnoses, contrasting sharply with 2–6% for late-stage diagnoses (Wang et al., 2020).
LUAD, representing 40% of lung cancer cases, affects both smokers and nonsmokers (Zappa and Mousa, 2016). It often originates from alveolar type 2 (AT2) cells, a type of alveolar epithelial cell, which tend to grow slowly at first but can spread early to distant organs (Sainz de Aja et al., 2021). AT2 cells secrete pulmonary surfactant, a lipoprotein complex that reduces surface tension in alveoli, and are crucial progenitors for maintaining proper alveolar integrity, lung structure, and function. Premature infants often lack mature AT2 cells, resulting in insufficient surfactant, which causes neonatal respiratory distress syndrome, a leading cause of premature newborn mortality.
At the molecular level, cancerous cells exhibit distinct alterations in gene expression and regulatory patterns governing critical cellular processes such as cell division, proliferation, signaling, DNA synthesis, and apoptosis (Motohara et al., 2019). For instance, epithelial cancer cells survive detachment by increasing the expression of histone demethylase KDM6B, which turns on genes like SOX2 and CD44 via epigenetic changes (Shait Mohammed et al., 2022). Noncoding RNAs, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), play pivotal roles in regulating gene expression through regulatory networks to maintain various biological and cellular processes.
LncRNA is a class of RNA molecules that exceeds 200 nucleotides in length and is devoid of protein-coding capacity. It regulates gene expression through diverse mechanisms, including interactions with miRNA and mRNA, and forms a competing endogenous RNA (ceRNA) regulatory network. Dysregulation in such networks has been implicated in various cancers (Qi et al., 2015; Xu et al., 2022). For example, the lncRNA GMDS-AS1 acts as a tumor suppressor by sequestering miR-96-5p, increasing CYLD mRNA expression associated with apoptosis (Zhao et al., 2020). In LUAD, reduced GMDS-AS1 levels diminish CYLD expression through the ceRNA network of the GMDS-AS1:miR-96-5p:CYLD axis, enhancing proliferation and reducing apoptosis. Conversely, lncRNA LCAT1 functions as an oncogene, promoting cell growth and development. Overexpression of this gene in lung cancer tissues is correlated with poor prognosis, likely through the sequestration of miR-4715-5p, which consequently upregulates the expression of its target RAC1 (Yang et al., 2019a). Several other lncRNAs have been reported in lung cancer, including the upregulation of HOTAIR, MALAT1, ANRIL, NEAT1, ZXF2, and HOTTIP, which are associated with a higher rate of metastasis and poor prognosis (Gencel-Augusto et al., 2023; Sun et al., 2017; Yang et al., 2015). Despite notable progress, our understanding of these complex regulatory networks and their biological functions in cancer remains incomplete.
The transition from AT2 cells to LUAD is characterized by significant pathophysiological changes that profoundly affect lung physiology and function. These changes disrupt pulmonary surfactant dynamics, alter alveolar structure, and compromise respiratory function in LUAD. The pulmonary surfactant primarily comprises 90% phospholipids and 10% proteins, including surfactant proteins SP-A, SP-B, SP-C, and SP-D (Carreto-Binaghi et al., 2016). Each of these proteins plays distinct biological roles crucial for maintaining pulmonary homeostasis. The synthesis of surfactant proteins is regulated by a set of specialized genes, including SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD.
Despite our current understanding of surfactant proteins and their genetic variants in different pulmonary diseases (Brudon et al., 2024; Lahti et al., 2004; Mitsuhashi et al., 2013; Sutton et al., 2022; Watson et al., 2020), the precise mechanisms underlying the alterations in the gene regulatory network governing surfactant gene expression in LUAD remain poorly understood, emphasizing the necessity for further investigation to enhance disease management. Therefore, our study aimed to elucidate the dysregulated expression of lncRNAs and their associated regulatory networks contributing to abnormalities in the pulmonary surfactant and respiratory systems in LUAD.
In this systems bioinformatics study, leveraging data from The Cancer Genome Atlas (TCGA) LUAD cohort, we identified differentially expressed mRNAs, lncRNAs, and miRNAs in LUAD. Subsequently, employing correlation coefficient analysis to examine the expression patterns of lncRNAs and mRNAs, we identified the downregulation of lncRNA SFTA1P and its coexpressed network. Intriguingly, this network appears to be associated with disrupting surfactant homeostasis and perturbing respiratory function in LUAD, thereby compromising lung physiology. Our findings contribute to a better understanding of the molecular mechanisms underlying surfactant homeostasis in LUAD and present a potential avenue for therapeutic interventions aimed at ameliorating lung function in affected individuals for more effective disease management.
Materials and Methods
The present study was conducted using publicly available data and did not require informed consent or research ethics board approval. The research presented here was performed under the overall research ethics oversight of the authors’ institutions.
Data curation
The RNA-seq, miRNA-seq, and clinical data of LUAD were retrieved from the Genomic Data Commons (GDC). The GDC serves as a repository for uniform genomic and clinical data from various cancer programs, including TCGA and Therapeutically Applicable Research to Generate Effective Treatments (TARGET). In this work, we used the Bioconductor (version 3.18) installed in the R environment (version 4.3.2). We used the GDCRNATools package (version 1.22.0) to download, integrate, and analyze data from GDC. Briefly, RNA-seq and miRNAs read counts data were downloaded using gdcRNADownload. The metadata associated with these files were parsed using gdcParseMetadata. The duplicated samples in RNA-seq metadata and miRNA metadata were filtered out using gdcFilterDuplicate. After that, we selected solid tissue normal (n = 59) and primary tumor (n = 516) sample types in RNA-seq metadata, while we selected solid tissue normal (n = 46) and primary tumor (n = 513) sample types in miRNA metadata using gdcFilterSampleType. Finally, raw counts data of RNA-seq and its metadata were merged into a single expression matrix using gdcRNAMerge. Similarly, raw count data of miRNA and its metadata were merged into a single expression matrix using gdcRNAMerge. Selecting a common “sample” between RNA-seq and miRNA metadata reduces the dataset to 20 solid tissue normal and 510 primary tumor sample types. Therefore, we analyzed differential expression separately for RNA-seq and miRNA-seq.
The raw count expression matrix of RNA-seq and miRNA-seq data was normalized separately using the gdcVoomNormalization function. The normalized RNA-seq and miRNA-seq data were used for Pearson correlation analysis.
Differential expression analyses of mRNAs, lncRNAs, and miRNAs
The raw count expression matrix of RNA-seq data was used to identify differentially expressed mRNAs (DEM) and differentially expressed lncRNAs (DEL) by the limma method with the gdcDEAnalysis and gdcDEReport functions. Similarly, the raw counts expression matrix of miRNA-seq data was used to identify differentially expressed miRNAs (DEMi) using the limma method with the gdcDEAnalysis and gdcDEReport functions. The differentially expressed mRNAs, lncRNAs, and miRNAs in cancer compared with normal were reported with |log2FC| > 1, and an adjusted p-value <0.01, corrected for the False Discovery Rate (FDR).
Functional enrichment analyses
In order to understand the biological roles and diseases correlated with differentially expressed mRNAs and lncRNAs, we performed the analysis for gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and disease ontology (DO) using the clusterProfiler and Disease Ontology Semantic and Enrichment (DOSE) packages. This step aimed to ascertain the biological processes, cellular components, and molecular functions significantly affected by the differentially expressed genes.
Construction of the regulatory network of lncRNA SFTA1P in LUAD
To build a coexpressed regulatory network of lncRNA SFTA1P, we selected 13 genes that showed a positive correlation with SFTA1P and were enriched across five distinct gene ontologies relevant to lung biological processes. Using the enrichment analysis tool, Enrichr-KG (https://maayanlab.cloud/enrichr-kg) with the modules: ARCHS4 transcription factors (TFs) Coexp, GO biological processes, and KEGG pathways, we identified a regulatory network, which we termed respiratory gaseous exchange and surfactant-associated Biological Network (rgsBNet).
Results
Differentially expressed mRNAs, lncRNAs, and miRNAs in LUAD
After removing duplicate samples, the final read count data of mRNAs and lncRNAs contained 575 samples (primary tumor = 516 and solid tissue normal = 59) and 60,616 genes of mRNAs and lncRNAs in LUAD. Similarly, the final data of miRNAs read count contained 559 samples (primary tumor = 513 and solid tissue normal = 46) and 2588 miRNAs in LUAD.
Our comprehensive analysis identified that 2987 protein-coding genes were differentially expressed (DEM) at |log2FC| > 1, and the adjusted p-value is <0.01. Among them, 1094 genes were upregulated, while 1893 genes were downregulated. This significant number of downregulated genes indicates the large-scale deactivation of specific pathways and functions in LUAD. The volcano plots show the differentially expressed genes in LUAD (Fig. 1A). The top 10 upregulated genes are FAM83A, CST1, PITX2, PRAME, CYP24A1, MMP13, COL11A1, MMP11, SYT12, and SPINK1; while the top 10 downregulated genes are SFTPC, CLDN18, LGI3, AGER, FABP4, GPM6A, UPK3B, SCGB1A1, CA4, and ANKRD1 (Table 1).
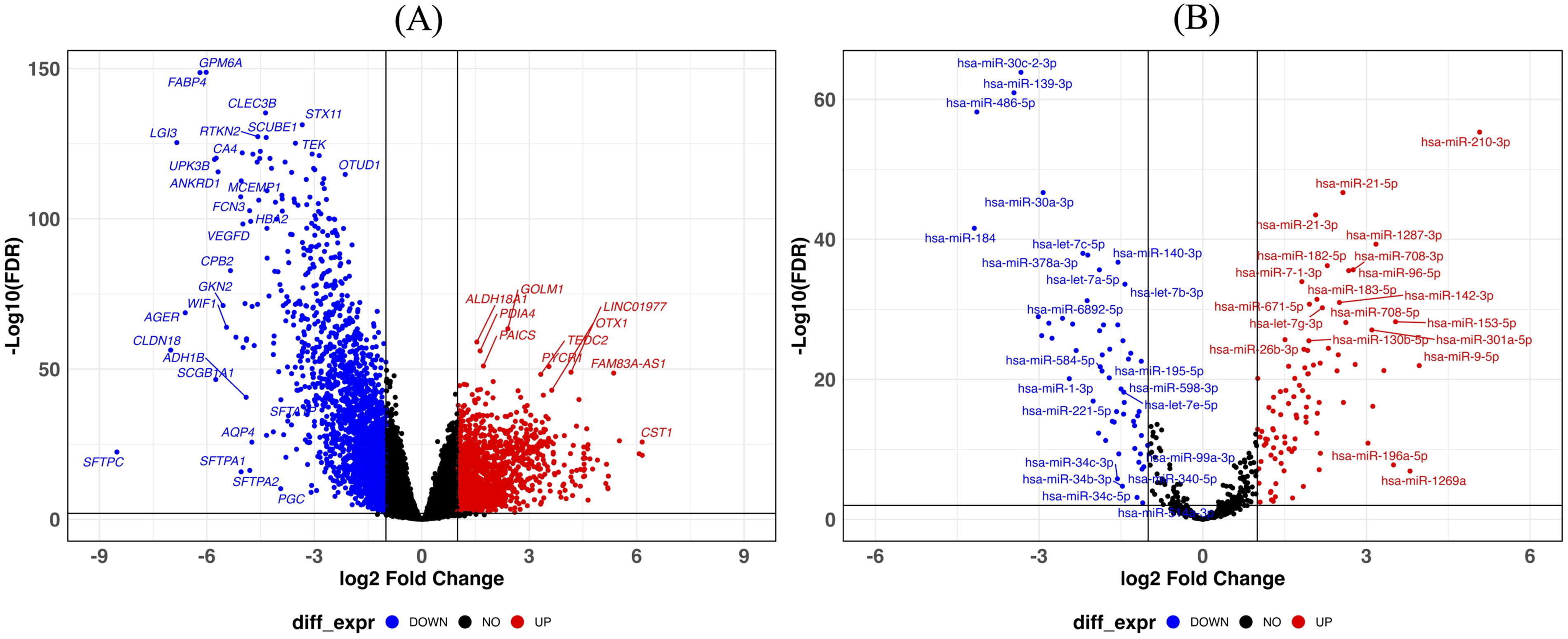
Volcano plot of mRNAs, lncRNAs, and miRNAs expression showing:
List of Top 10 Upregulated and Downregulated mRNAs, lncRNAs, and miRNAs in LUAD Samples Compared with Normal Samples
lncRNAs, long noncoding RNA; LUAD, lung adenocarcinoma; miRNAs, microRNAs.
In addition, we identified 209 differentially expressed lncRNAs between LUAD and normal tissue samples. Of these, 115 were upregulated, whereas 94 were downregulated (Fig. 1A). The top 10 upregulated lncRNAs are LCAL1, FAM83A-AS1, AFAP1-AS1, Z98257.1, FEZF1-AS1, MNX1-AS1, AC008496.3, ZFPM2-AS1, LINC01977, and AL391056.1; while top 10 downregulated lncRNA are AC008268.1, FENDRR, LHFPL3-AS2, LRRK2-DT, SFTA1P, AC009244.1, AL121933.2, LINC01936, PELATON, and HHIP-AS1 (Table 1).
Further analysis revealed 150 differentially expressed miRNAs, 93 of which miRNAs were upregulated, whereas 57 were downregulated (Fig. 1B). The prevalence of upregulated miRNAs alongside downregulated mRNA indicates a shift in miRNA-mediated gene silencing patterns, where miRNAs possibly prevent the expression of genes involved in cellular and extracellular matrix stability, promoting cell proliferation. The top 10 upregulated miRNAs are hsa-miR-210-3p, hsa-miR-9-5p, hsa-miR-1269a, hsa-miR-153-5p, hsa-miR-196a-5p, hsa-miR-3607-3p, hsa-miR-1287-3p, hsa-miR-577, hsa-miR-301a-5p, and hsa-miR-31-5p; while the top 10 downregulated miRNAs are hsa-miR-184, hsa-miR-486-5p, hsa-miR-139-3p, hsa-miR-30c-2-3p, hsa-miR-144-5p, hsa-miR-1247-3p, hsa-miR-30a-3p, hsa-miR-144-3p, hsa-miR-451a, and hsa-miR-133a-3p (Table 1). The list of all DEM, DEL, and DEMi results is provided in Supplementary Tables S1, S2, S3, S4, S5 and S6.
Functional enrichment of DEM and DEL
Furthermore, we examined the functional enrichment of DEM and DEL to understand the underlying biological processes and molecular mechanisms affected during LUAD progression.
Upregulated genes
According to biological processes of GO, the upregulated genes showed prominent involvement in chromosome segregation, nuclear division, and DNA replication (Fig. 2A). Thus, these genes are important for proper cell division, and their aberrations can result in uncontrolled cell division and cancer development. Regarding the cellular components of GO, we found that these gene products are localized within chromosomal regions and spindles (Fig. 2B). This specific positioning reinforces their implication in cell division and strongly implies their participation in uncontrolled cell proliferation, a key inducer of tumorigenesis. In the context of molecular functions of GO, these genes exhibited associations with essential cellular activities, including ATP hydrolysis and catalytic activity on DNA (Fig. 2C). These functions are integral to cellular energy metabolism, a process known to be elevated in cancer cells. Consequently, these pathways are viable candidates for targeted therapeutic interventions aimed at curbing the heightened metabolic activity characteristic of cancer.

Enrichment analysis of genes upregulated in LUAD:
Considering DO, correlations emerged between the upregulated genes and various cancer types, including breast carcinoma, ovarian cancer, and LUAD (Fig. 2D). This noteworthy overlap suggests shared molecular mechanisms across these cancers and indicates potential cross-cancer therapeutic strategies. These commonalities present a possible avenue for developing targeted interventions with broader applicability. With the KEGG pathways analysis, we uncovered that those upregulated genes were involved in critical cellular processes, such as the cell cycle, motor proteins, and amino acids biosynthesis (Fig. 2E). The heightened activity of the cell cycle pathway underscores its role as a key target in cancer therapy. Furthermore, the upregulation of amino acid biosynthesis reflects the increased demand for protein synthesis, which is a critical requirement for rapid cell growth and division of cancer cells. Supplementary Tables S7, S8, S9, S10 and S11 provide the complete list of enrichment results for upregulated mRNAs and lncRNAs.
Downregulated genes
According to biological processes of GO, the downregulated genes have roles in cell-substrate adhesion and leukocyte migration (Fig. 3A). The downregulation of cell-substrate adhesion, which is vital for precise cellular localization and migration, may contribute to the loss of tissue architecture, a hallmark feature of LUAD. Furthermore, the observed impairment in leukocyte migration suggests a dampening of the immune response, potentially providing the tumor with a means to evade immune protection. These alterations may converge to create a microenvironment conducive to tumor progression and metastasis. Regarding the cellular components, a substantial proportion of the downregulated genes constitute the collagen-containing extracellular matrix (Fig. 3B). Collagen, crucial for structural integrity, provides vital support to tissues. The downregulation of these genes may lead to altered cell morphology and tumor microenvironment, ultimately facilitating invasive growth and metastasis. In molecular functions, these genes are significantly involved in essential cellular activities, particularly actin binding and carbohydrate binding (Fig. 3C). Actin, an important cytoskeletal protein, maintains cellular structure, motility, and integrity. Carbohydrate-binding proteins are crucial for mediating cell−cell communication. The downregulation of these functions may potentially lead to cellular disarray, resulting in unregulated cell growth and impaired communication—characteristic features of cancer cells.
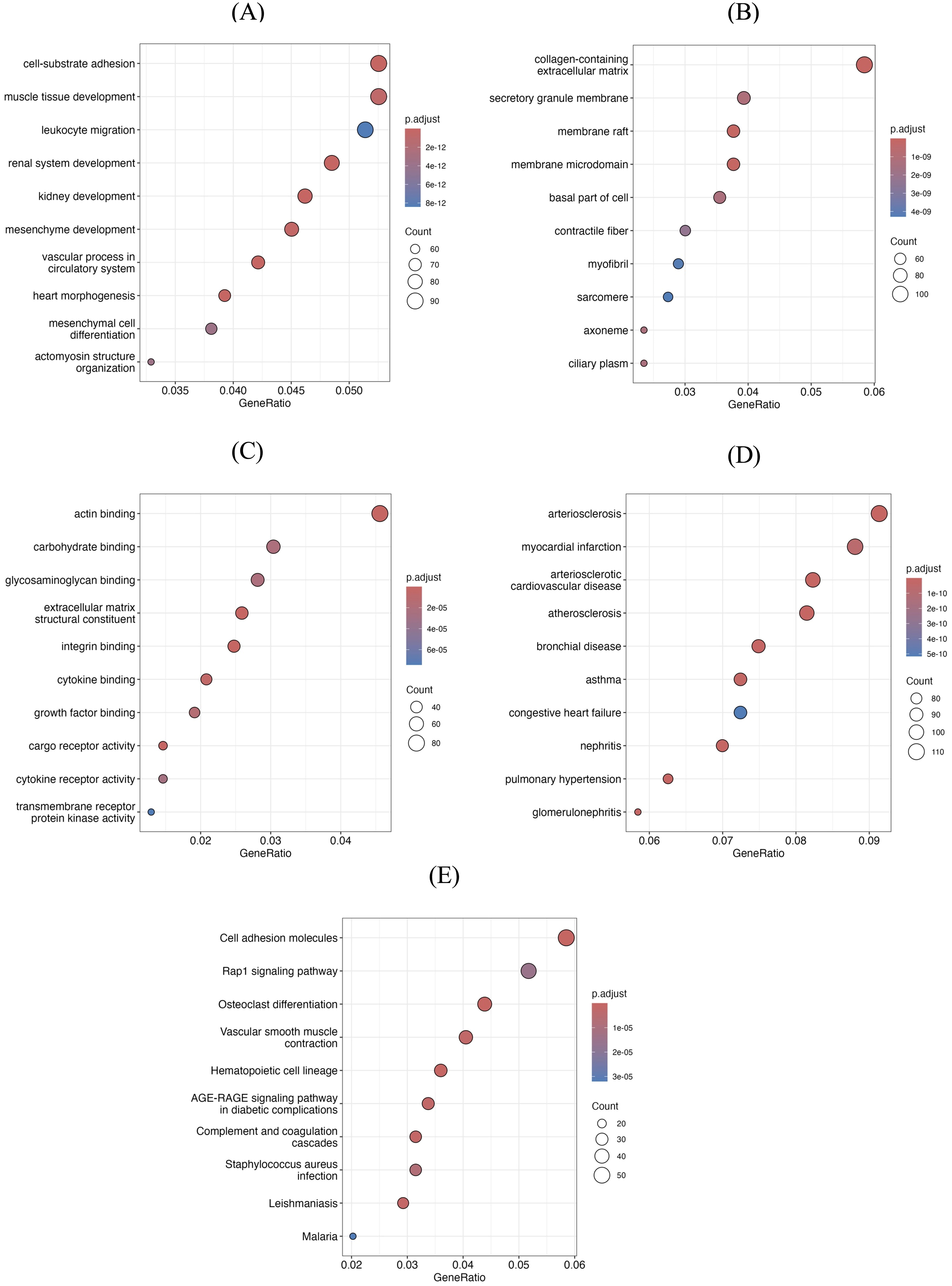
Enrichment analysis of genes downregulated in LUAD:
Further analysis of DO revealed associations between downregulated genes in LUAD and conditions such as arteriosclerosis, myocardial infarction, and bronchial disease, indicating potential shared molecular mechanisms underlying these conditions (Fig. 3D). Regarding the KEGG pathways enrichment analysis, downregulated genes were prominently featured in pathways related to cell adhesion molecules and Rap1 signaling (Fig. 3E). Impairments in cell adhesion may lead to the loss of contact inhibition—a recognized hallmark of cancer. Additionally, disruptions of Rap1 signaling can exert extensive effects on various cellular processes, ranging from cell growth to apoptosis. The complete list of enrichment results for downregulated mRNAs and lncRNAs is provided in Supplementary Tables S12, S13, S14, S15 and S16.
Expression correlations between lncRNAs and mRNAs in LUAD pathophysiology
We investigated the correlation between lncRNAs and mRNAs expression in LUAD to uncover key regulatory mechanisms underlying disease progression and possible therapeutic targets. Our study identified 4706 distinct correlations between 123 lncRNAs and 1215 protein-coding genes, using a correlation cutoff > |0.5| and a p-value <0.05 (Supplementary Table S17). Among these lncRNAs, SFTA1P shows correlations with 245 protein-coding genes (Fig. 4A). While SFTA1P expression is most prominent in normal lung tissues, it undergoes significant downregulation in LUAD, a pattern that intensifies with disease progression to advanced stages (Fig. 4B–D). Although the downregulation of SFTA1P in lung cancer has been reported previously, the molecular mechanisms have not been clearly elucidated (Du et al., 2020). Consequently, to unravel the regulatory network and understand the impact of SFTA1P on LUAD pathophysiology, we explored the enrichment analysis of protein-coding genes highly correlated with SFTA1P.
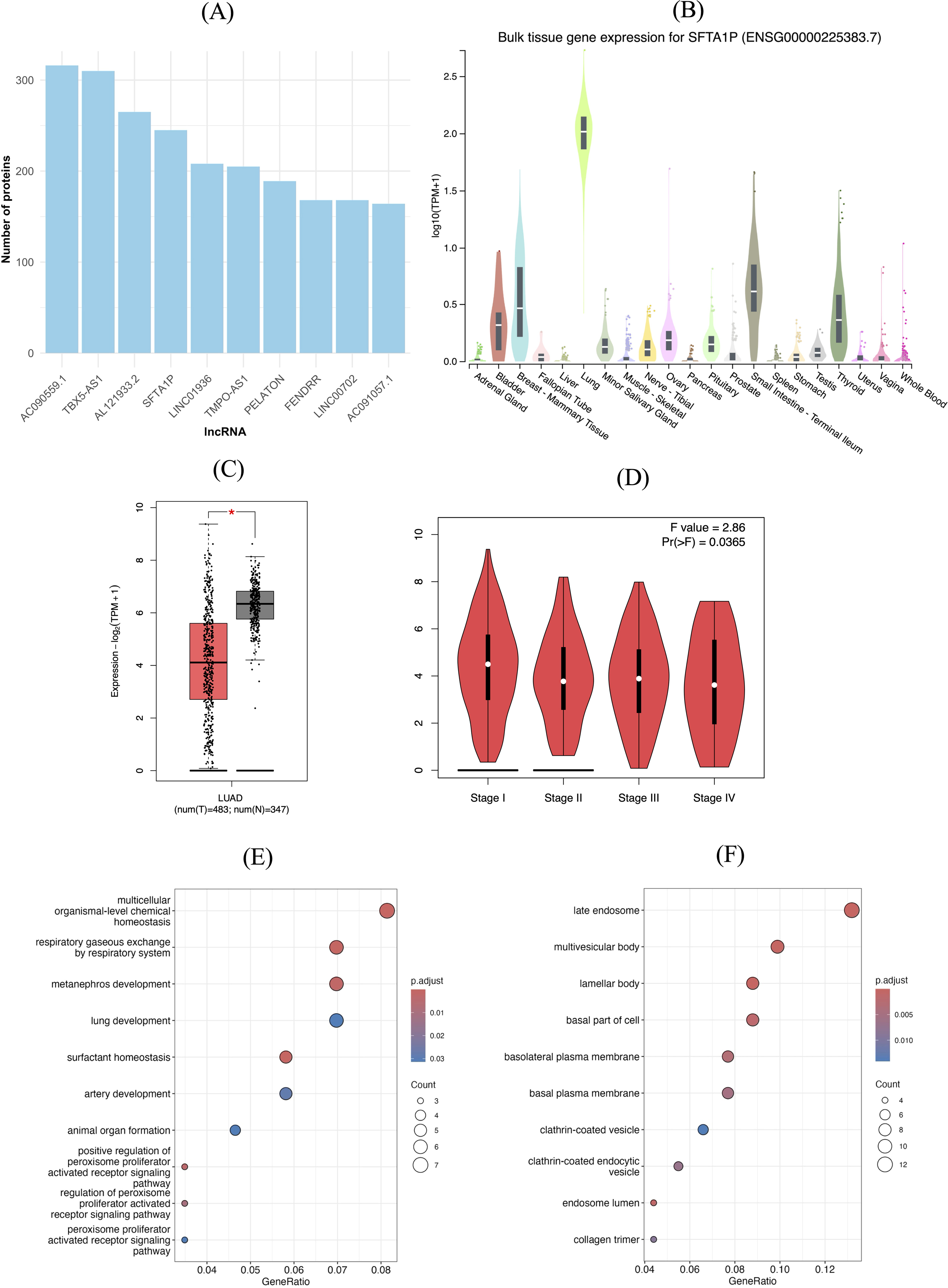
Functional enrichment of SFTA1P correlated genes
Negatively correlated genes with downregulated SFTA1P enhance cell division in LUAD
The 152 genes exhibiting a negative correlation with SFTA1P were upregulated in LUAD. These genes were strongly associated with essential biological processes such as chromosome segregation, nuclear division, and sister chromatid segregation (Supplementary Fig. S1A). The cellular components of GO analysis revealed that these genes are located at the chromosome region, spindle, and kinetochore, indicating their enhancement in critical processes such as DNA replication and segregation during cell division (Supplementary Fig. S1B). In molecular functions analysis of GO, negatively correlated genes have roles in tubulin binding, microtubule binding, ATP hydrolysis activity, and helicase activity (Supplementary Fig. S1C). Furthermore, the upregulated genes were associated with different cancers and cell cycle pathways, emphasizing their contribution to enhancing cell proliferation and cell division (Supplementary Fig. S1D and E).
Genes positively correlated with downregulated SFTA1P compromise respiratory gaseous exchange and surfactant homeostasis in LUAD
Our investigation identified 94 downregulated genes having positive correlations with SFTA1P and have the potential to identify gene regulatory network associations with crucial biological processes in LUAD. Enrichment analysis of biological processes revealed their involvement in respiratory gaseous exchange by the respiratory system, lung development, and surfactant homeostasis (Fig. 4E). Furthermore, these genes reveal their association with cellular components, including the late endosome, multivesicular body, lamellar body, clathrin-coated vesicles, and endosome lumen, which are important for surfactant formation and vesicle transport (Fig. 4F). Thus, the accumulating evidence indicates that the downregulation of SFTA1P and its positively correlated genes in LUAD compromise surfactant homeostasis in the lungs and respiratory function. Therefore, the restoration of SFTA1P function potentially holds promise for improving lung surfactants, which are essential for alveolar stability, lung physiology, respiratory function, and therapeutic strategies for LUAD.
The regulatory network governing surfactant homeostasis and respiratory function in LUAD
In our continued investigation, we aimed to elucidate the complex regulatory network governing dysregulated respiratory function and surfactant homeostasis in LUAD. To achieve this goal, we conducted an analysis of 13 genes that exhibited enrichment across five distinct gene ontologies relevant to lung biological processes. These genes displayed a positive correlation with SFTA1P, along with the presence of two TFs, TCF21, and HOPX (Table 2). Utilizing enrichment analysis tools, such as ARCHS4 TFs Coexp, GO biological processes, and KEGG pathways, through Enrichr-KG (https://maayanlab.cloud/enrichr-kg) at default parameters, we unraveled a comprehensive regulatory network, termed rgsBNet. This network revealed the intricate interactions among genes, TFs, and enriched biological processes, providing possible underlying molecular mechanisms in LUAD.
Selected Gene Ontology Biological Process Terms for Genes That Display Positive Correlations with the lncRNA SFTA1P Were Used to Construct the Coexpression Regulatory Network rgsBNet
rgsBNet, respiratory gaseous exchange and surfactant-associated biological network; GO, gene ontology.
Within the rgsBNet, five important TF genes—NKX2-1, EPAS1, NR4A1, TBX4, and TCF21—emerged as key regulators, suggesting their possible roles in modulating the expression of genes crucial for respiratory gaseous exchange and surfactant homeostasis (Fig. 5A). The genes in rgsBNet are intricately linked to crucial biological processes governing lung development and function, including surfactant homeostasis, tissue chemical homeostasis, and respiratory tube development. Additionally, pathway enrichment analysis revealed associations with cellular homeostasis and signaling pathways such as phagosome, pertussis, lysosome, ABC transporters, and the Hedgehog signaling pathway (Supplementary Table S18).
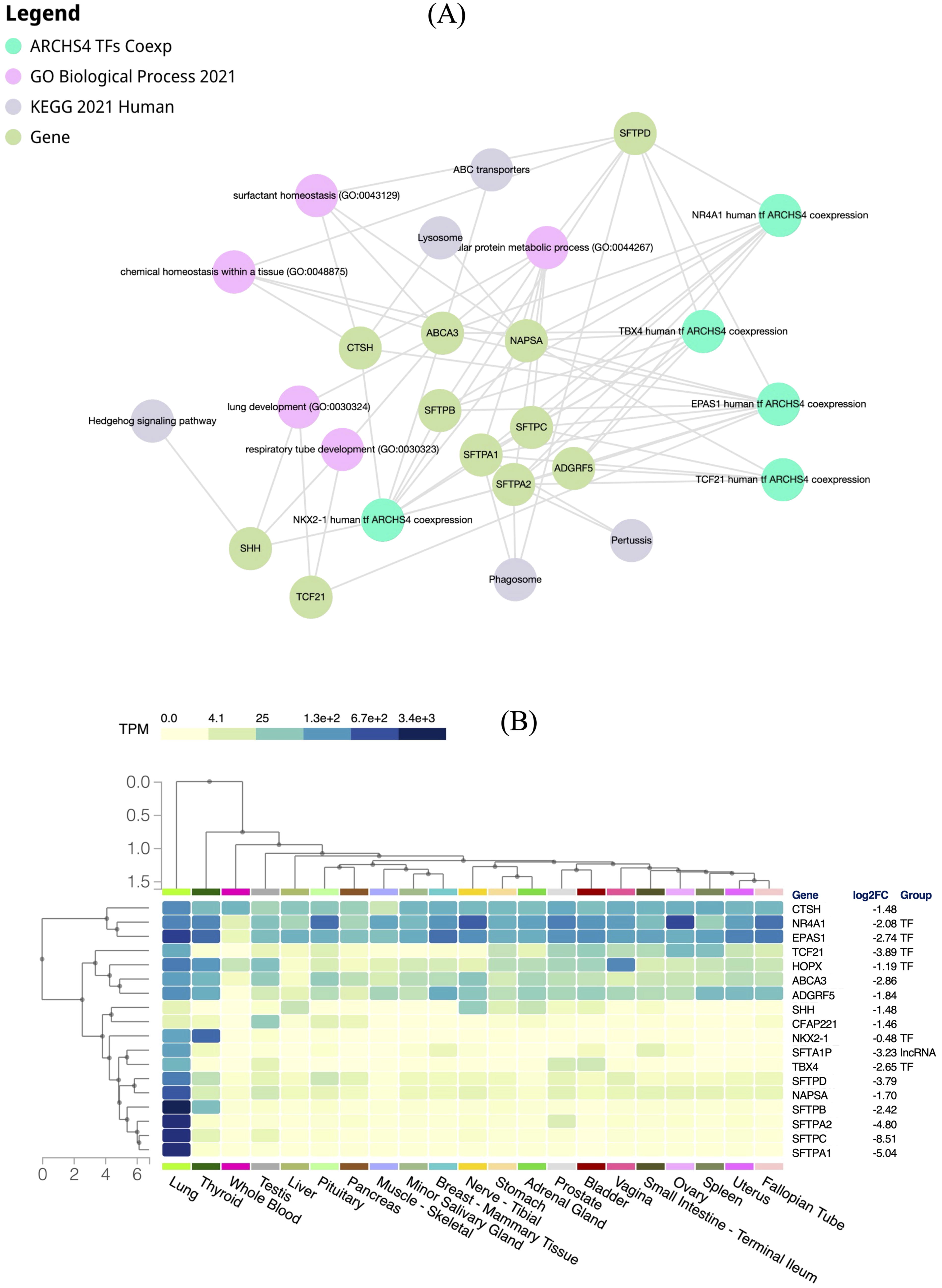
The downregulation of SFTA1P and other genes within the rgsBNet network in LUAD suggests potential regulatory mechanisms underlying disease pathogenesis. The first hypothesis considers the potential role of the aberrant function or expression of TFs in modulating downstream targets within the rgsBNet network. Therefore, we also checked the expression level of these genes in normal lung tissue using the GTEx portal (https://gtexportal.org). Our analysis showed that all genes in rgsBNet were downregulated in LUAD compared with normal samples and were termed as signature genes (Fig. 5B). However, most of them are highly expressed in normal lung tissue than in other human tissues (Fig. 5B).
The differential analysis of the signature genes within rgsBNet revealed significant downregulation of these genes in LUAD (Supplementary Fig. S2). A similar downregulation was observed in LUSC, another form of NSCLC (Supplementary Fig. S2). However, no significant differences were noted in other cancers during pan-cancer analysis across TCGA datasets (Supplementary Fig. S2). Another plausible mechanism involves downregulated SFTA1P acting as a molecular “sponge” for certain miRNAs, sequestering them away from their target mRNAs. Consequently, reduced expression of SFTA1P could lead to increased availability of miRNAs to bind and suppress the expression of their target mRNAs in the rgsBNet network. Thus, the identification of key regulatory elements and their functional associations provides valuable insights into the pathogenesis of LUAD, thereby offering potential targets for therapeutic intervention and biomarker discovery.
To unravel the intricate regulatory mechanisms, we systematically investigated the available data of experimentally validated miRNA-SFTA1P interactions. Our analysis using DIANA tools (https://diana.e-ce.uth.gr/lncbasev3/interactions) revealed that two miRNAs, hsa-miR-15a-5p and hsa-miR-17-5p, interact with SFTA1P (Supplementary Fig. S3). Further analysis demonstrated an upregulation of hsa-miR-17-5p in LUAD compared with normal tissue and downregulation of hsa-miR-15a-5p in LUAD (Supplementary Tables S5 and S6). These findings indicate that decreased SFTA1P levels in LUAD may lead to increased hsa-miR-17-5p activity due to reduced sequestration, although experimental validation is required to confirm this interaction.
Correlation between SFTA1P expression and immune infiltrates in LUAD
We further investigated the relationship between SFTA1P expression and immune cell infiltration in LUAD using the TIMER2 tool (http://timer.comp-genomics.org/), with adjustments for tumor purity (Li et al., 2020). The analysis revealed significant associations between SFTA1P expression and various types of immune infiltration, highlighting its potential role in modulating the tumor immune microenvironment. A heatmap (Fig. 6A) shows the correlation between SFTA1P expression and immune infiltration levels across pan-cancer from TCGA data. Notably, the scatter plots provided more specific insights into the relationship in LUAD. SFTA1P expression exhibited a significant negative correlation with T cell CD4+ Th2 infiltration (Fig. 6B) and myeloid-derived suppressor cells (MDSCs) (Fig. 6C), both with correlation coefficients below −0.5 (p-value <0.05). These findings indicate that lower SFTA1P expression is associated with induced infiltration of Th2 cells and MDSCs in LUAD, potentially contributing to the immunosuppressive environment that facilitates tumor progression.

Association between immune infiltrates and SFTA1P expression in LUAD, as analyzed using the TIMER2 tool with purity adjustment.
Conversely, SFTA1P expression showed a positive correlation with hematopoietic stem cells (HSCs) (Fig. 6D), with a correlation coefficient above 0.5. This positive association indicates that lower levels of SFTA1P expression in LUAD may lead to a reduction in HSCs within the tumor microenvironment. This association could potentially influence tumor growth and immune response because HSCs play a role in regulating immune cell production and function. Overall, these results suggest that SFTA1P significantly affects the immune landscape of LUAD. By modulating the infiltration of various immune cell types, SFTA1P may play a crucial role in tumor biology and immune evasion mechanisms. These findings highlight the multifaceted role of SFTA1P in LUAD and underscore the need for further studies to explore its potential as a therapeutic target or biomarker for improved cancer management (Arias et al., 2021; Gabrilovich and Nagaraj, 2009; Hiam-Galvez et al., 2021; Kumar and Goyal, 2021; Passegué and Weisman, 2005; Somasundaram and Herlyn, 2009).
Discussion
LUAD remains one of the most prevalent and lethal cancers. Therefore, unraveling the molecular mechanisms underlying LUAD is crucial for identifying new therapeutic targets and developing effective treatment strategies. LUAD often originates from the transformation of AT2 cells, which play a critical role in producing pulmonary surfactants and maintaining alveolar structure and function. The disruption of surfactant homeostasis and respiratory function in patients with LUAD poses significant challenges in patient management. Previous research has extensively studied the roles of surfactant proteins and their encoding genes in various lung diseases (Gower and Nogee, 2011; Magnani and Donn, 2020; Whitsett and Weaver, 2015). However, the regulatory networks governing surfactant production and respiratory function in LUAD are not well understood, which hinders the translation of scientific discoveries into clinical applications.
Recent studies underscore the importance of multiomics data in analyzing gene regulatory networks to understand disease pathogenesis and identify therapeutic targets in various cancers, including lung cancer. For example, our previous study proposed that the TFs FOXM2 and MYBL2 may contribute to NSCLC progression by regulating key downstream genes involved in cell proliferation (Ahmed, 2019). Similarly, Liu et al. demonstrated the role of MYBL2 in bladder cancer by interacting with FOXM1 and activating the Wnt/β-catenin signaling pathway (Li et al., 2020). In addition, multiomics and gene regulatory networks have been used to identify biomarkers for cancer diagnosis and prognosis. For example, gene expression and protein−protein interaction data with machine learning (ML) were employed to develop a highly accurate predictive model for NSCLC (Ahmed et al., 2022). Another study used CpG site methylation as a prognostic epigenetic signature in high-grade gliomas (Drexler et al., 2024). Recently, we used multiomics and ML to identify transcriptomics and epigenetic signatures and developed a computational model for predicting hepatocellular carcinoma with high accuracy (Ahmed et al., 2024).
Multiomics analysis of gene regulation in LUAD
Our current study utilized multiomics data to investigate gene regulation in LUAD, focusing on the dysregulated network underlying surfactant homeostasis and respiratory function. Our current study provides a comprehensive analysis of differentially expressed mRNAs, lncRNAs, and miRNAs in LUAD, revealing significant dysregulation that contributes to the disease’s molecular landscape. This analysis identified 2987 DEM in LUAD, with 1094 upregulated and 1893 downregulated (Fig. 1A, Table 1). Notably, the upregulation of genes such as FAM83A and CST1 and the downregulation of SFTPC and CLDN18 align with previous findings showing their association with cancer progression and poor prognosis (Braicu et al., 2019; Dai et al., 2017; Sanada et al., 2006; Zhou et al., 2023; Zuo et al., 2023).
Functional enrichment and pathway analysis in LUAD
Functional enrichment analysis of the upregulated genes revealed their roles in chromosome segregation, nuclear division, and DNA replication (Fig. 2). These processes are essential for cell proliferation, and their dysregulation is a hallmark of cancer, as noted by Hanahan and Weinberg (Hanahan and Weinberg, 2011). The association of these genes with cancer-related pathways, such as the cell cycle and amino acid biosynthesis (Fig. 2), underscores their significance in LUAD pathogenesis. Conversely, downregulated genes were enriched in processes such as cell-substrate adhesion, leukocyte migration, collagen-containing extracellular matrix, and actin binding (Fig. 3). This downregulation implies a compromise in structural integrity and impaired cellular communication, facilitating tumor invasion and metastasis, consistent with prior studies on the role of the extracellular matrix and immune evasion in cancer progression (Junttila and de Sauvage, 2013).
LncRNA SFTA1P and its correlated gene enrichment
Our investigation identified SFTA1P (log2FC = −3.23, FDR = 6.80E-40) as significantly downregulated in LUAD. A previous study also showed that SFTA1P downregulates in NSCLC and acts as a tumor suppressor through inhibiting the phosphatidylinositol 3-kinase–protein kinase B (PI3K-AKT) signaling pathway and inducing cell cycle arrest (Du et al., 2020). Interestingly, our correlation analysis showed SFTA1P has interactions with 245 protein-coding genes (Fig. 4A). The functional enrichment of genes negatively correlated with SFTA1P indicated their involvement in cell division and proliferation (Supplementary Fig. S1), supporting the hypothesis that SFTA1P downregulation enhances cell proliferation in LUAD. This finding aligns with other research implicating lncRNAs in cell cycle regulation across various cancers (Chen et al., 2023b; Fang et al., 2022; Tokgun et al., 2020; Yin et al., 2023). Accumulating evidence suggests that SFTA1P acts as a tumor suppressor, as evident by an earlier study (Du et al., 2020). Thus, our analysis indicates that SFTA1P and its negatively correlated genes, which are enriched for proliferation pathways, warrant further investigation as potential therapeutic targets in LUAD.
LncRNA SFTA1P implications of surfactant production in LUAD
The genes positively correlated with SFTA1P were enriched in processes related to respiratory gaseous exchange and surfactant homeostasis (Fig. 4E–F). Their downregulation in LUAD indicates compromised lung function and surfactant production, which are important for maintaining alveolar stability and respiratory efficiency. Surfactants, lipid-protein complexes produced within the lungs, serve as the linchpin for the lung’s structural and functional integrity including reducing alveolar surface tension. This reduction ensured that the alveoli did not collapse during exhalation, facilitating consistent and efficient gaseous exchange. In addition, the lungs function as a first line of defense, blocking and neutralizing airborne pathogens to protect the respiratory system.
Dysregulation of surfactant proteins is a hallmark of lung cancer, contributing to the disease’s pathogenesis and progression (Watson et al., 2020; Whitsett and Weaver, 2015). Notably, altered expression of surfactant proteins, such as SFTPA, SFTPB, SFTPC, and SFTPD, is frequently observed in LUAD (Fig. 5); however, the role of SFTA1P in their regulation remains unclear. Given its expression in AT2 cells and potential tumor-suppressive effects, SFTA1P may directly or indirectly modulate surfactant protein production, possibly via miRNA interactions, TFs, or signaling pathways.
Our study identified the regulatory network rgsBNet, which encompasses genes and TFs associated with surfactant homeostasis and respiratory function (Fig. 5A). The downregulation of these genes in LUAD and their high expression in normal lung tissue highlight their critical role in lung biology (Fig. 5B). Similar regulatory networks have been described in the context of lung development and disease, indicating conserved mechanisms across different contexts (Barnes et al., 2019; Borek et al., 2023; Miller and Spence, 2017; Samarelli et al., 2021). The significant downregulation of these genes in patients with both LUAD and LUSC suggests shared molecular mechanisms across NSCLC subtypes (Supplementary Fig. S2). Although the precise mechanistic role of SFTA1P in the etiology of LUAD remains to be elucidated, its association with surfactant production presents a compelling avenue for further investigation. There is a plausible hypothesis that perturbations in surfactant dynamics, influenced by the SFTA1P gene, might lead to the formation of a pulmonary microenvironment conducive to tumorigenesis. Such alterations could compromise alveolar structural integrity, thereby facilitating tumor cell invasion.
To validate the role of SFTA1P, two complementary approaches will be considered. First, knockdown of SFTA1P in primary human AT2 cells using small interfering RNAs (siRNAs), with efficiency validated by quantitative real-time PCR (qRT-PCR) (Gao et al., 2018). Followed by an assessment of its impacts on surfactant protein expression and oncogenic traits, including proliferation, migration, and reduction of apoptosis will be assessed (Zhang et al., 2017). Second, the overexpression of SFTA1P in the A549 cell line, delivered through lipid nanoparticles (LNPs), will be evaluated for its effects on surfactant production and tumor suppression, which will be monitored using similar assays (Zhang et al., 2017).
SFTA1P downregulation in LUAD might be a secondary effect rather than a driver event, potentially caused by dysregulated TFs, mutations, or epigenetic changes (Shait Mohammed et al., 2022). Therefore, verifying its driver versus passenger status requires whole-genome sequencing (WGS) and bisulfite sequencing to detect genomic and epigenetic alterations at the SFTA1P promoter (Nazari et al., 2025; Rheinbay et al., 2017). Additionally, chromatin immunoprecipitation sequencing (ChIP-seq) targeting TFs from our study (Fig. 5B) could elucidate SFTA1P downregulation mechanisms in LUAD (Chen et al., 2016).
A prior study showed that miR-17-92 cluster overexpression in a transgenic mouse model promotes the proliferation of lung epithelial progenitor cell and inhibits its differentiation, partly via miR-17-5p targeting Rbl2 (Lu et al., 2007). However, Lu et al. noted that no single miR-17-5p target explains the lung developmental phenotype, implying the involvement of multiple targets. Our study indicates hsa-miR-17-5p and hsa-miR-15a-5p potentially target SFTA1P (Supplementary Fig. S3). The upregulation of hsa-miR-17-5p and downregulation of SFTA1P in LUAD suggest that SFTA1P fails to effectively sequester these miRNAs, thereby inducing oncogenic effects. This is consistent with the established role of hsa-miR-17-5p as an oncogenic miRNA in various cancers (Bobbili et al., 2017; Stoen et al., 2021). However, these interactions require experimental validation via RNA pull-down assays using biotinylated SFTA1P probes incubated with AT2 or A549 cell lysates and streptavidin bead capture (Torres et al., 2018). Small RNA sequencing can confirm bound miRNAs (e.g., oncogenic hsa-miR-17-5p and other miRs). Furthermore, investigating shared TFs that regulate these identified miRNAs could inform the development of more effective therapeutic strategies.
LncRNA SFTA1P and immune infiltration
We examined the relationship between SFTA1P expression and immune cell infiltration in LUAD using TIMER2. We observed that reduced SFTA1P expression was associated with higher infiltration of T cell CD4+ Th2 and MDSCs (Fig. 6B and C), which might contribute to an immunosuppressive tumor microenvironment and promote tumor progression (Chen et al., 2023a; Liu et al., 2022; Nakamura and Smyth, 2020; Yang et al., 2019b). Conversely, SFTA1P expression was positively correlated with HSCs, suggesting a potential role in immune modulation (Fig. 6D). These findings underscore the multifaceted role of SFTA1P in LUAD, which affects both tumor growth and immune evasion. This finding aligns with studies that highlighted the impact of the tumor microenvironment and immune infiltration on cancer progression (Yang et al., 2019b). Moreover, altered tumor microenvironment in LUAD—potentially associated with SFTA1P downregulation—may promote neoplastic cell proliferation and immune cell infiltration, thereby contributing to tumor progression (Abbaszadegan et al., 2017; Junttila and de Sauvage, 2013; Rhim, 1989). Our results emphasize the need for further research to validate SFTA1P as a therapeutic target or biomarker for improved cancer management.
Future prospects of SFTA1P and its clinical implications
SFTA1P’s critical role in LUAD suggests promising prognostic and therapeutic potential. Therefore, the expression level of SFTA1P in LUAD biopsies, measured by qRT-PCR or in situ hybridization, may serve as a prognostic biomarker, with low expression potentially indicating a more aggressive disease (Fig. 4D). Therapeutically, restoring SFTA1P expression using synthetic RNA-based constructs—mimicking mRNA vaccine technology (Pardi et al., 2018) and delivered via LNPs—represents a promising approach. The efficacy and safety of this strategy can be validated in LUAD models. Additionally, further investigation into the study of SNPs in SFTA1P and their association with LUAD could provide valuable insights into disease susceptibility and progression.
Our findings provide insights into targeted therapies for LUAD. Future research should focus on the functional characterization of the identified genes and RNAs through both in vitro and in vivo studies to validate these findings. Prioritizing understanding of SFTA1P’s role in surfactant regulation and its effects on lung physiology may inform targeted therapeutic strategies and improve patient outcomes.
Limitations of this study
Although our study offers valuable insights based on bioinformatics analysis of publicly available data, future studies must validate these findings using in vitro and in vivo LUAD models to enable translation into clinical applications. Therefore, the proposed mechanisms underlying the observed phenotypes and potential therapeutic interventions remain hypothetical until experimental confirmation.
Conclusions
In this systems bioinformatics study, we report that the disruption of the lncRNA SFTA1P network is consistent with impairing surfactant homeostasis and respiratory function and warrants attention for future drug discovery and development. Employing a multidimensional analysis integrating lncRNAs, miRNAs, and mRNAs, our investigation identified pivotal regulators and interconnected pathways critical for LUAD pathogenesis. Our findings focused on the possible role of SFTA1P and its regulatory network—including associated proteins and upstream TFs—in modulating surfactant production and regulating lung function, which are perturbed in LUAD.
This finding underscores the therapeutic potential of SFTA1P, highlighting its promise as a target for restoring surfactant homeostasis and preserving pulmonary function in patients with LUAD. Building upon these insights, future research should incorporate a multidisciplinary approach—including in vivo models, in vitro systems, and clinical investigations—to further elucidate the functional role of SFTA1P and translate these findings into innovative diagnostic and therapeutic strategies for LUAD.
Footnotes
Acknowledgment
The authors would like to thank the University of Jeddah for its technical and financial support.
Authors’ Contributions
F.A. is the principal investigator who conceptualized and designed the project, collected and analyzed data, interpreted the results, and wrote and revised the article. Y.M.R. evaluated and interpreted the results and wrote and revised the article.
Author Disclosure Statement
The authors declare that there is no competing interest.
Funding Information
This work was funded by the University of Jeddah, Jeddah, Saudi Arabia, under Grant No. (UJ-23-DR-97).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.