
Abstract
Idiopathic pulmonary fibrosis (IPF) and pulmonary hypertension (PH) are two chronic conditions that can coexist occasionally, resulting in high morbidity and mortality. Despite their clinical association, the underlying genetic mechanisms and therapeutic targets that might link these two chronic disorders remain poorly understood. The present study used in silico analyses and machine learning to uncover genetic features and potential therapeutic targets shared by IPF and PH. Differentially expressed genes (DEGs) were identified using RNA sequencing data from the Gene Expression Omnibus, which revealed a total of 13 common DEGs between IPF and PH. Importantly, among the identified genes, TFF3 was significantly upregulated in both diseases. TFF3 is targeted by aminoglutethimide as identified through the Drug Gene Interaction Database, and with an interaction score of 3.26. Using the Protein Contact Atlas, PROCHECK, PROSA, and ProtParam tools, the structural model of TFF3 was validated. Finally, molecular docking analysis demonstrated a binding affinity score of −6.1 kcal/mol between TFF3 and aminoglutethimide, indicating a stable and potentially effective interaction between aminoglutethimide and the target protein. Aminoglutethimide displayed favorable ADMET properties as well. In conclusion, this in silico study reports (1) potential overlapping molecular links between IPF and PH, and (2) in silico potential of aminoglutethimide and TFF3 in drug repurposing for therapeutic interventions targeting both IPF and PH. These findings also challenge the traditional paradigm of pharmaceutical innovation that has long relied on the “one drug, one disease” premise, and highlight the potentials of “one drug, polydisease” paradigm of drug discovery and development.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive interstitial lung disease (ILD) characterized by rapid scarring or fibrosis of the lung parenchyma, leading to a decline in respiratory function (Richeldi et al., 2017). The prevalence of IPF varies globally, ranging from 7 to 1650 cases per 100,000 individuals, with higher rates observed in older populations (Shah Gupta et al., 2023). Diagnosis is often challenging and typically involves high-resolution computed tomography scans, and in some cases, lung biopsies to be able to ascertain the hallmark patterns of the disease.
IPF frequently coexists with comorbidities such as cardiovascular diseases, gastroesophageal reflux disease, and pulmonary hypertension (PH), which significantly worsen symptoms and clinical outcomes of the disease (Caminati et al., 2019; Dasgupta, 2025a). Among these, PH is particularly severe, as it often worsens breathlessness, limits exercise capacity, and is associated with reduced survival. PH is characterized by elevated blood pressure in the pulmonary arteries, which, if left untreated, can progress to right heart failure (Choudhury et al., 2021; Collum et al., 2017). The cross talk between IPF and PH is mediated through overlapping mechanisms such as vascular remodeling, chronic inflammation, and hypoxia-driven signaling cascades. It is well known that both diseases share common hallmarks such as chronic inflammation, Transforming Growth Factor-beta (TGF-β) signaling, oxidative stress, and endothelial-to-mesenchymal transition (EndMT) (Ruffenach et al., 2020). In PH, structural alterations affect all layers of the vascular wall, with key involvement of endothelial and smooth muscle cells (Graham et al., 2011). In IPF, fibroblast foci and disrupted alveolar architecture dominate the pathology (Yamashita et al., 2009). Identifying common genetic features between IPF and PH is crucial, as it may uncover novel molecular targets for therapeutic intervention (Alharbi, 2024; Collum et al., 2017; Ruffenach et al., 2020).
The reported prevalence of PH among patients with IPF varies widely, especially among those evaluated for lung transplantation. Studies have described PH in 20–46% of IPF patients undergoing right heart catheterization (RHC) as part of transplant assessment (Patel et al., 2007; Rivera-Lebron et al., 2013; Shorr et al., 2007). Notably, Nathan et al. (2008) demonstrated that PH prevalence increased from 38.6% at initial transplant evaluation to 86.4% at the time of transplantation, suggesting that disease progression and timing of diagnostic assessment critically influence detection rates (Nathan et al., 2008). Furthermore, a population-based study from the United States in 2018 reported 30,335 hospitalizations with a diagnosis of IPF, of which 8075 (26.6%) were also diagnosed with PH (Desai et al., 2024). These data reflect the significant clinical burden and demographic disparities in the coexistence of IPF and PH.
Despite the clinical association between IPF and PH, the underlying genetic mechanisms and therapeutic targets that might link these two chronic disorders remain inadequately understood. Hadi et al. (2023) reported the potential of saracatinib and sotatercept in managing IPF associated with PH (Hadi et al., 2023). Rajagopal et al. (2021) reviewed the molecular mechanisms linking lung fibrosis and vascular remodeling, in support of the development of targeted pharmacotherapies. Waxman et al. (2022) discussed diagnostic and therapeutic advances in PH associated with ILDs, highlighting the significance of RHC in diagnosis. Collum et al. (2017) explored the high prevalence of PH in IPF patients and the associated mortality risk, calling for deeper insights into molecular mechanisms and treatment strategies. Gaikwad et al. (2020) delved into EndMT as a crucial mechanism connecting vascular remodeling and fibrosis, proposing it as a potential therapeutic target. Collectively, these studies underscore the urgent need for innovative approaches to understand and manage the intricate relationship between IPF and PH.
Bioinformatic-based research has identified several genetic targets and predictive drug candidates that may be effective for the treatment of conditions that can occasionally overlap such as IPF and PH (Barrett et al., 2013; Dasgupta, 2025b). Studies have also used detailed structural investigation of the identified important proteins to understand their physicochemical and stereochemical properties (Hossain et al., 2022; Tripathi et al., 2024). Bioinformatic tools such as ProtParam can be used to evaluate the physicochemical properties of common proteins implicated in both IPF and PH, while the Protein Contact Atlas provides structural information. Furthermore, PROCHECK tool aids in the assessment of stereochemical quality of the target protein (Kayikci et al., 2018). In silico docking studies involving these shared proteins and potential therapeutic compounds offer critical insights into drug–protein interactions. Molecular docking helps predict the binding affinity and orientation of drugs within the active sites of target proteins, aiding in the rational design of precision therapeutics.
The present study leverages bioinformatic analyses and tools as noted above to uncover the potential genetic overlaps between IPF and PH, aiming to deepen our understanding of the molecular interconnections underlying these diseases. A predictive machine learning model, built on the basis of altered genes, evaluated their significance as potential disease biomarkers. The expression of these genes was validated using independent datasets to ensure robustness. In addition, an in silico analysis highlighted a key protein and its interacting drug molecule, with their interaction confirmed through molecular docking analysis. A detailed structural snapshot of the target protein and the drug candidate provided further insights into its functional dynamics, potentially informing the future design of drug repurposing and precision medicine strategies.
Materials and Methods
Identification of the DEGs in IPF and PH
The data harnessed in this study were obtained from the publicly available and accessible National Center for Biotechnology Information-Gene Expression Omnibus (NCBI-GEO) database (http://www.ncbi.nlm.nih.gov/geo/) and did not require research ethics board approval or informed consent. The study and analyses reported herein were conducted under the overall research ethics oversight of the author’s institution.
Using the keywords “idiopathic pulmonary fibrosis” and “pulmonary hypertension,” relevant datasets were identified and screened. Datasets were selected based on the following criteria: (1) human-derived samples, (2) relevance to IPF or PH, (3) inclusion of both patient and healthy control samples, and (4) availability of raw or processed RNA-seq data. This study incorporated the datasets GSE231693 and GSE236251, which provide insights into gene expression changes in IPF and PH compared with controls, respectively.
The GSE231693 dataset comprises bulk RNA sequencing data generated from lung tissue samples of 20 IPF patients and 18 healthy controls. Sequencing was performed using the Illumina HiSeq 4000 platform (Jia et al., 2023). The dataset was developed by Genentech from explanted lung tissues collected during transplantation from patients diagnosed with ILD, including systemic sclerosis-associated ILD, and IPF. The broader cohort included a total of 208 subjects with a mean age of 48.09 years (standard deviation: 12.43). The median forced vital capacity percentage predicted was 82.48, with an interquartile range of 71.12–93.30. For the present study, a subset of this cohort was selected, consisting of 20 IPF patients and 20 healthy controls.
The GSE236251 dataset comprises RNA sequencing data obtained from pulmonary artery trunk samples collected during heart transplantation. The original cohort for this dataset consisted of four groups as follows: donor controls (n = 33), patients with left heart disease without PH (LHD w/o PH; n = 41), patients with PH-LHD (n = 49), and patients with pulmonary arterial hypertension (n = 4). Demographic characteristics showed that the donor group included 10 females and 23 males with a median age of 41 years (range: 16–72). The PH-LHD group included 14 females and 35 males with a median age of 55 years (range: 31–65). Median body weights varied across subgroups, with females in the PH-LHD group averaging 65 kg (range: 50–91) and males 85 kg (range: 55–127). For the purpose of the present analysis, pulmonary artery trunk samples from patients with PH-LHD (n = 18) and healthy donor controls (n = 4) were included to ensure direct disease-relevant comparison. RNA was extracted using the TRIzol method following snap-freezing of the arterial tissue in liquid nitrogen. Sequencing was conducted using the Illumina HiSeq 2000 platform (Kucherenko et al., 2023). The overall study design of the present study is shown in Figure 1.

Workflow of the study design.
Screening of common DEGs
The GEO2R package was used to analyze RNA sequencing data from the GSE231693 and GSE236251 datasets. The updated definitions provided by GEO2R for sample and control groups were followed during dataset analysis (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html#groups). This tool uses the limma and GEOquery algorithms to perform gene expression analyses, identifying DEGs with rigorous control over multiple testing. The significant DEGs were initially screened using a raw p value threshold of <0.05. Subsequently, gene expressions meeting the criteria of |log2(fold change)| > 2 and an adjusted p value <0.05 were selected for further analysis. The adjusted p values were calculated using the Benjamini–Hochberg method to control for false discovery rate). This two-step filtering approach ensures robust statistical significance while minimizing type I errors that are common in high-throughput RNA-seq analyses.
Validation of the common genes using independent dataset
The expression levels of the identified DEGs were validated using additional datasets obtained from the NCBI-GEO repository. Two datasets, GSE79544 and GSE188938, were selected for this validation process. The GSE79544 dataset includes RNA-seq transcriptomic profiles from 7 normal smoking controls and 16 IPF smoking patients, based on RNA extracted from macrophages isolated from bronchoalveolar lavage fluid (Lee et al., 2018). The GSE188938 dataset comprises 12 peripheral blood samples, including 7 samples from patients with chronic thromboembolic PH (CTEPH), a subtype of PH, and 5 samples from healthy individuals (Xu et al., 2022a). Validation in these biologically distinct but disease-relevant tissues further supports the robustness and cross-cohort consistency of the DEGs identified in the discovery datasets.
Machine learning model for biomarker candidate discovery
The machine learning approach was used to evaluate the potential of the common DEGs as disease biomarkers using the MetaboAnalyst web tool (Pang et al., 2021; Xia et al., 2009). The random forest algorithm was applied to assess the predictive value of the genes, with the analysis conducted using 500 trees and 7 predictors. The algorithm was implemented with 500 trees, as this is the default and widely accepted setting in the MetaboAnalyst platform. This configuration is supported by the foundational work of Breiman (2001) and Liaw and Wiener (2002), who demonstrated that increasing the number of trees improves classification stability until reaching a performance plateau (Breiman, 2001; Liaw and Wiener, 2002). This approach is also consistent with established practices in omics-based random forest applications (Pawloski et al., 2022; Pink, 2016). Feature importance plots were generated to rank the significance of each gene in distinguishing disease states.
Correlation and comparison between gene expression in IPF and PH
The correlation between gene expression in IPF and PH was analyzed. A heatmap was generated to visualize the relationships between gene expression levels across the two conditions. Pearson correlation coefficients were used to quantify the strength and direction of the associations. Darker blue indicates negative correlation, whereas the red color suggests positive correlation between the genes of IPF and PH subjects. The expression levels of the genes were also compared between IPF and PH using GraphPad Prism version 8.0.1. The bar chart plots showed the expression of these DEGs in both cases.
Drug–gene interaction identification
The Drug Gene Interaction Database (DGIdb) was used to identify potential molecular drug candidates that could target the commonly dysregulated genes in IPF and PH. DGIdb (https://www.dgidb.org/search_interactions) is a comprehensive resource for drug–gene interactions, containing information about human drugs, “druggable genes,” and various drug–gene interactions from multiple sources. At present, it includes over 14,144 drug–gene interactions covering 2611 human genes and 6307 drugs (Wagner et al., 2016).
Absorption, distribution, metabolism, excretion, and toxicity studies
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis is essential for assessing the pharmacodynamic properties of potential drug candidates. This analysis is based on the ligand SMILES, obtained via the PubChem tool, and provides a structured approach to evaluating drug viability. This approach aids in the systematic assessment of drug-likeness and safety profiles of the identified potential drug from DGIdb, thereby supporting the selection of promising therapeutic candidates with favorable ADMET characteristics (Ferreira and Andricopulo, 2019).
Toxicity and bioavailability prediction
The toxicity levels of the predicted drugs were thoroughly examined using the Protox-II webtool (https://tox-new.charite.de/protoxII/). Protox-II uses computational models trained on real in vitro and in vivo data to estimate the toxicity potential of both existing and virtual compounds. For each input compound, the tool calculates the acute toxicity class and various endpoints by analyzing chemical similarities to known toxic substances and applying these trained models (Banerjee et al., 2018). The toxicity analysis results provide the predicted median lethal dose (LD50) in mg/kg body weight, along with the toxicity class and prediction accuracy for each drug compound analyzed.
Homology modeling
To assess the homology modeling of the target protein, the corresponding Protein Data Bank (PDB) ID was retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB-PDB) database and analyzed using the SWISS-MODEL tool (https://swissmodel.expasy.org/) (Waterhouse et al., 2018). Model accuracy was evaluated using the PROCHECK tool (https://saves.mbi.ucla.edu/). A high-quality model is characterized by an ERRAT quality factor exceeding 50 and more than 80% of residues achieving a score ≥0.2 in the 3D/1D profile, as determined by the VERIFY3D server (Gundampati et al., 2012). The atomic composition and amino acid profile of the target protein is evaluated using ProtParam tool (https://web.expasy.org/protparam/). The quality of modeled structures was also validated by ProSA tool (https://prosa.services.came.sbg.ac.at).
Molecular docking
Preparation of protein and ligand
The target protein was prepared following the methodology described by Tallei et al. (2020). For this study, the 3D crystal structure of the target receptor was retrieved from the RCSB-PDB (http://www.rcsb.org/pdb/).
Similarly, the 3D structures of the selected drugs were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/compound) and were downloaded in a simplified data format for further processing.
Docking of ligands into protein
Molecular docking was carried out using AutoDock Vina integrated with UCSF-Chimera© version 1.13 software (Trott and Olson, 2010). For receptor preparation, water molecules were removed, and polar hydrogen atoms and Gasteiger charges were added. The ligand structures were also processed by incorporating hydrogen atoms and charges. The processed protein and ligand structures were saved as PDBQT files before initiating the docking procedure.
The docking process generated 10 configuration files, representing the top 10 docking poses for each protein–ligand complex. The pose with the lowest binding energy (kcal/mol) and minimal root mean square deviation (RMSD) was selected as the optimal docking configuration, indicating the strongest and most favorable interaction.
Results
Identification of the common DEGs in IPF and PH
In case of IPF, 871 upregulated and 211 downregulated genes were observed compared with the controls. A comparative analysis of gene expression between PH and controls revealed a total of 245 elevated and 59 decreased genes in the patient group. Comparative analysis revealed 13 DEGs with consistent expression trends in both diseases, suggesting potential shared molecular mechanisms. Among these, ADAMTS18, TFF3, MUC16, PPP1R1A, SYT12, HCAR1, C14orf180, DNAH9, and C6 were significantly upregulated, whereas RNASE2, CNTN6, SIGLEC10, and SIGLEC10-AS1 were consistently downregulated in both IPF and PH, compared with controls.
Validation of common genes
The majority of the 13 common DEGs appeared to exhibit similar expression patterns across independent validation datasets (GSE79544 and GSE188938), thereby supporting the observations made during the discovery phase. This consistency may suggest that these DEGs play a reproducible role in the molecular landscape shared by IPF and PH, highlighting their potential relevance for further investigation as diagnostic or therapeutic targets.
Machine learning model analysis
Random forest, a popular machine learning model, was developed to distinguish IPF and PH from controls based on the expression of 13 common genes. The model’s performance was evaluated using the out-of-bag (OOB) error. For IPF classification, the model achieved an OOB error of 0.154. For PH classification, the model demonstrated improved performance with an OOB error of 0.105. These results suggest that the random forest model effectively differentiates both disease groups from controls, with particularly high accuracy in classifying PH cases. These results demonstrate the model’s strong potential in distinguishing IPF and PH from controls, with varying levels of classification accuracy across the groups (Supplementary Fig. S1).
Correlation and comparison between IPF and PH
A heatmap was generated to visualize the correlation between IPF and PH using 13 DEGs, with blue indicating negative correlations and red representing positive correlations close to 1. The analysis revealed strong positive correlations among several genes, such as PPP1R1A and C14orf180 (0.952966), as well as SIGLEC10 and SIGLEC10-AS1 (0.988131). Moderate positive correlations were observed between CNTN6 and RNASE2 (0.546617), as well as HCAR1 and C6 (0.80817). Negative correlations were prominent between PPP1R1A and genes such as CNTN6 (−0.25324) and RNASE2 (−0.20742). Similarly, C14orf180 showed a negative correlation with CNTN6 (−0.2259) and RNASE2 (−0.16939) (Supplementary Fig. S2). These findings highlight intricate gene interactions, providing insights into the molecular underpinnings shared between IPF and PH.
Upon direct comparison between IPF and PH, it was observed that PPP1R1A and C14orf180 showed a higher expression in PH than in IPF. In contrast, C6, DNAH9, RNASE2, TFF3, HCAR1, SIGLEC10, SYT12, MUC16, ADAMTS18, and SIGLEC10-AS1 exhibited a relatively higher expression in IPF compared with PH. These findings support the outline of the disease-specific transcriptional signatures and provide additional insights into the pathobiological differences between IPF and PH. The expression profiles of these genes in the IPF versus PH group are illustrated in Supplementary Figure S3.
Structural analysis of potential drug
A total of 36 drugs targeting the 13 common DEGs were identified using the DGIdb database, of which 11 are currently approved for clinical use. The complete list of drug–gene interactions, along with their approval status by the Food and Drug Administration (FDA) and the corresponding DGIdb interaction scores, is provided in Supplementary Table S1. Among these, aminoglutethimide was selected for further analysis as it is an FDA-approved drug, its chemical structure is available in the PubChem database, and it targets TFF3, one of the overlapping genes identified between IPF and PH in this study (interaction score: 3.26). Aminoglutethimide was further evaluated through ADMET analysis to assess its pharmacokinetic properties and toxicity profile as a potential therapeutic candidate targeting TFF3. Its chemical formula is C13H16N2O2, with a molecular weight of 232.28 g/mol and 17 heavy atoms. Aminoglutethimide passes all key pharmacological filters, including Lipinski’s, Ghose, Veber, Egan, and Muegge rules, with a bioavailability score of 0.55. Toxicity analysis shows an LD50 value of 1800 mg/kg, classifying it as toxicity class 4 (low toxicity), suggesting that aminoglutethimide could be a viable candidate for further investigation in the treatment of IPF and PH (Fig. 2).
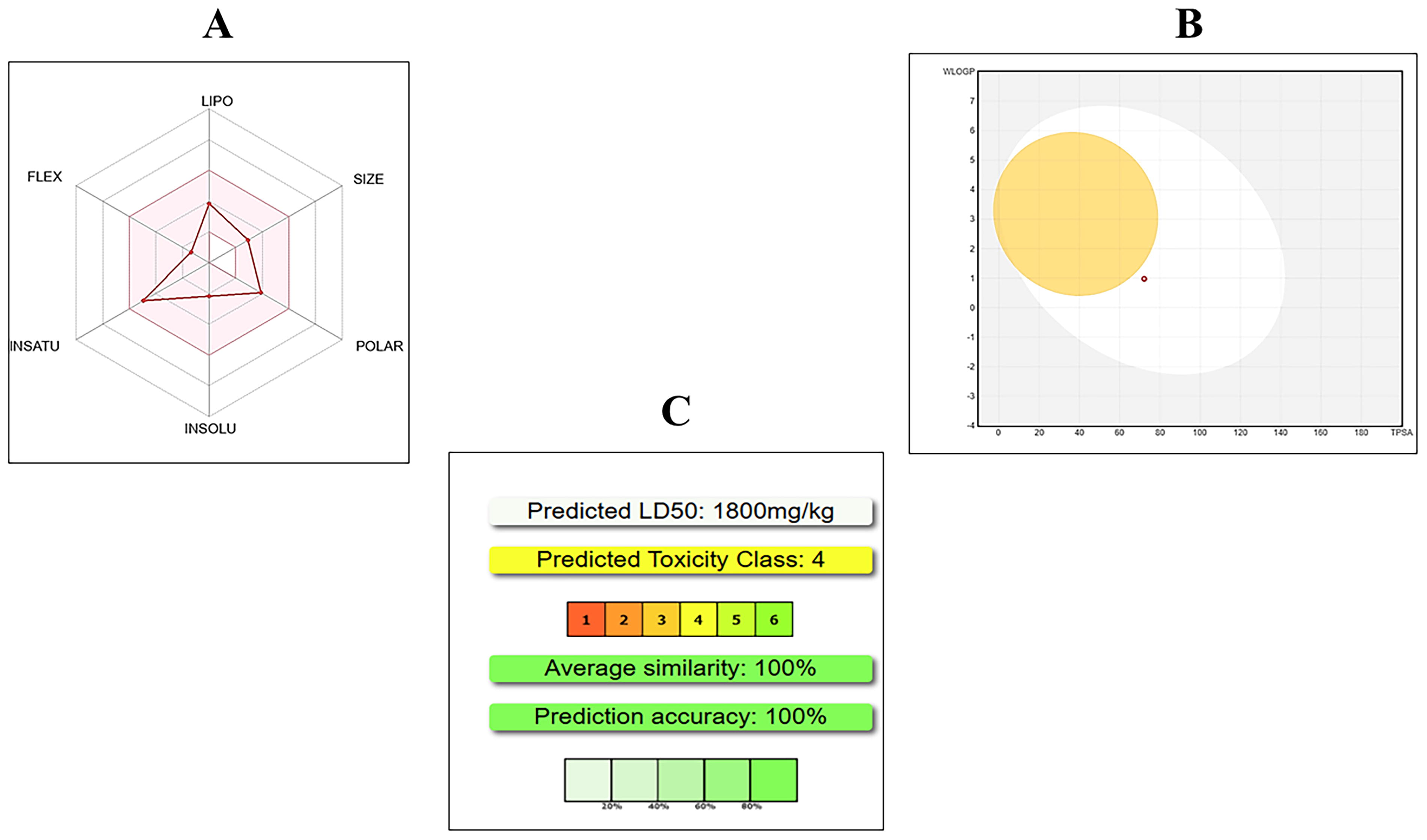
Structural validation and composition analysis of TFF3
The structural model of TFF3 was validated using Protein Contact Atlas, PROCHECK, PROSA, and ProtParam tools. Ramachandran Plot analysis revealed 90.2% of residues in the most favored regions, 9.8% in additional allowed regions, and none in generously allowed or disallowed regions, indicating a high-quality model. G-factor scores for dihedral angles and main-chain covalent forces were −0.15 and 0.43, respectively, with an overall average score of 0.09, reflecting good stereochemical properties. The PROSA Z-score of −5.62 further confirmed the structural quality compared with high-resolution crystal structures (Fig. 3). ProtParam analysis determined the atomic composition as C374H589N103O112S10, with 1188 atoms in total. The amino acid composition revealed high proportions of alanine (11.2%), leucine (10%), and cysteine (10%), along with other residues such as glycine (6.2%) and proline (7.5%) (Fig. 4A). Finally, the structural and interaction insights of the TFF3 protein based on Protein Contact Atlas analysis are shown in Figure 4B–D. The validation results and composition metrics demonstrate that the TFF3 structural model is of reliable quality and suitable for further computational and experimental analyses.
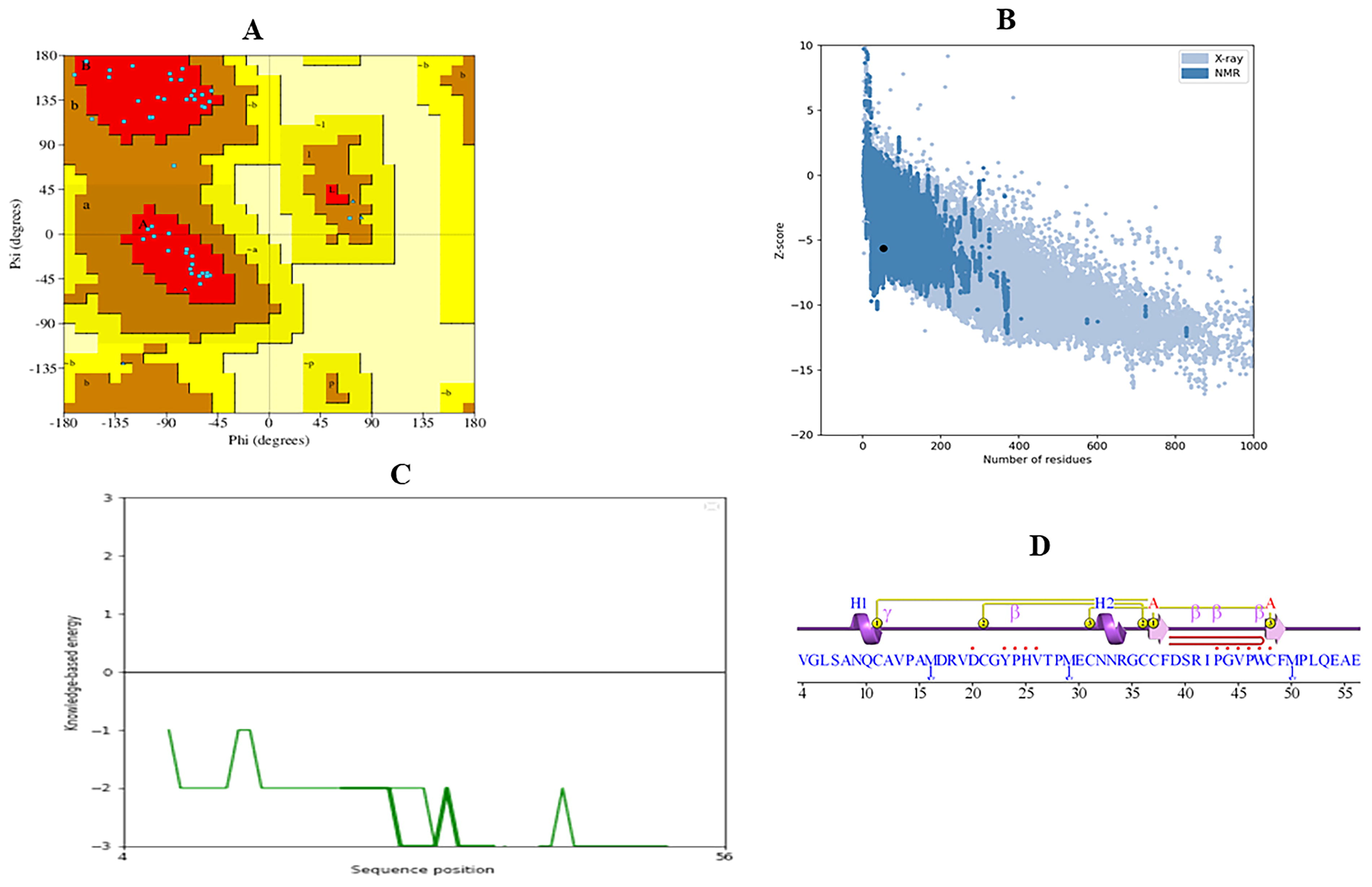
Validation of the TFF3 structural model.
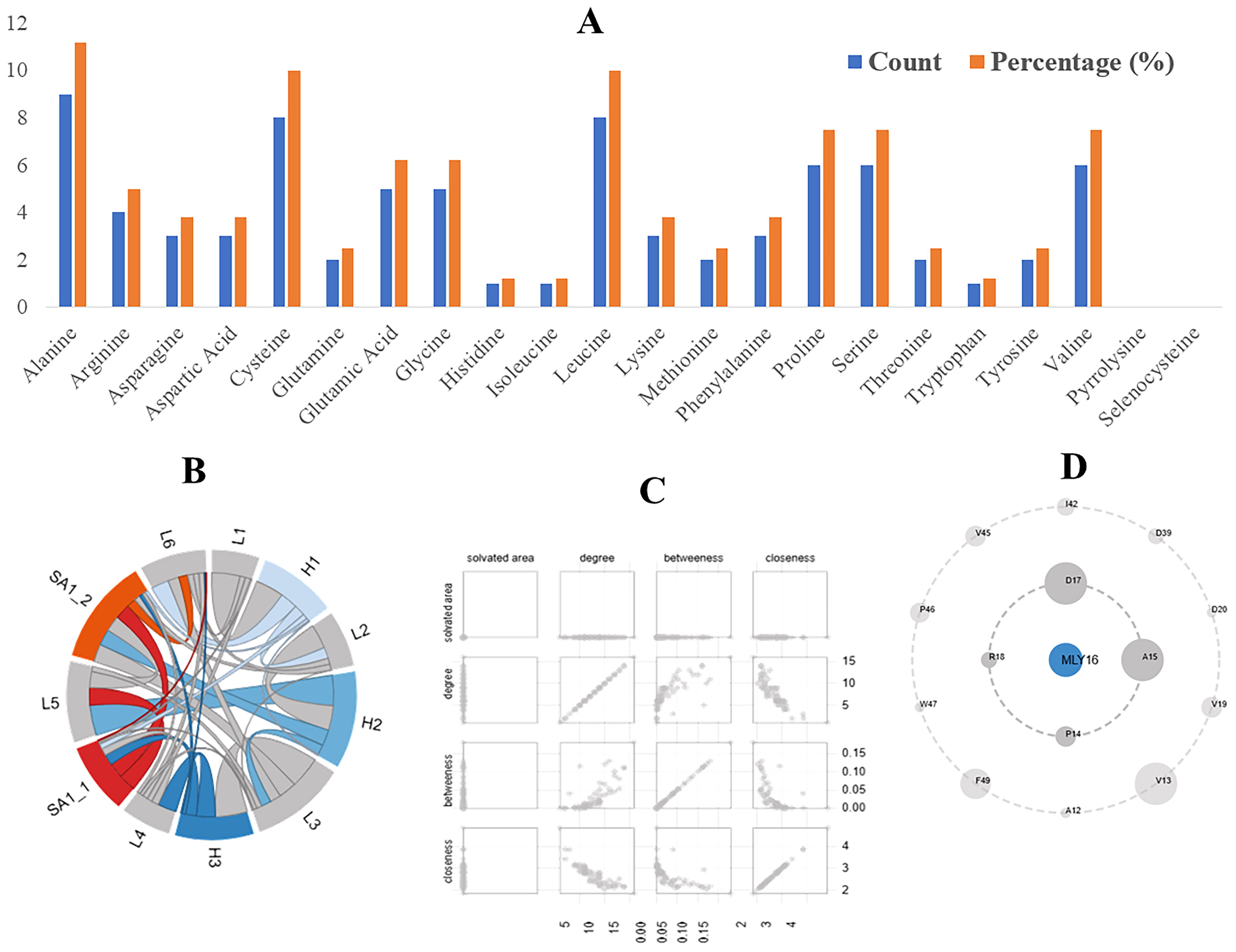
Structural and interaction analysis of the TFF3 protein using in silico tools.
Molecular docking analysis
Molecular docking analysis was performed to assess the binding affinity between aminoglutethimide and TFF3 protein. The binding site of the TFF3 protein was identified and visualized using UCSF Chimera. The docking procedure was performed using AutoDock Vina, with the grid box defined around the predicted binding domain. Default grid parameters were applied, centered on the largest surface-accessible pocket of the modeled structure. The grid box was centered at coordinates (X: 4.23, Y: 12.90, Z: −10.16) with dimensions (Size X: 44.44, Y: 46.18, Z: 53.07) to encompass the entire active site region. The final docking run generated multiple binding poses, and the best pose was selected based on binding energy and RMSD criteria. The docking analysis demonstrated a binding affinity score of −6.1 kcal/mol, indicating a stable and potentially effective interaction between the drug and the target protein (Fig. 5). A lower binding affinity score generally reflects a stronger and more favorable interaction between the ligand (aminoglutethimide) and the receptor (TFF3).

Molecular docking results of aminoglutethimide and TFF3. Binding pose of aminoglutethimide within the active site of the TFF3 protein. The interaction features a binding affinity score of −6.1 kcal/mol, indicating strong ligand–receptor interaction.
Discussion
Drug repurposing and thinking beyond the disease silos in drug design, discovery, and clinical development are among the contemporary approaches in pharmaceutical innovation (Dasgupta et al., 2023; Dasgupta, 2024a, 2024b;). Uncovering the molecular substrates shared among two or more diseases can help develop interventions with a broad therapeutic and diagnostic scope. The overlap in the genetic signatures between IPF and PH observed in the present study highlights a potential convergence in their molecular mechanisms, which may help explain their frequent coexistence in clinical settings.
The identification of 13 DEGs with consistent expression trends in both diseases suggests a shared pathogenic framework that could be driven by chronic inflammation, fibrosis, and vascular remodeling. Genes such as ADAMTS18, TFF3, and MUC16, which were significantly upregulated as observed in the present analyses, are associated with processes such as extracellular matrix remodeling, epithelial integrity, and immune response modulation (Chen et al., 2024; Meyer zum Büschenfelde et al., 2006; Nie et al., 2024). Similarly, the consistent downregulation of genes such as RNASE2 and CNTN6 points toward disrupted cellular communication and immune regulation (Acquati et al., 2019). This shared gene expression profile suggests that common environmental triggers, such as hypoxia or inflammatory insults, might initiate or exacerbate pathological changes in both diseases. Supplementary Figure S4 illustrates the possible involvement of common genetic features, cellular components, and pathways that may potentially contribute to the pathogenesis of both IPF and PH.
The performance of the random forest model in distinguishing IPF and PH from controls using these 13 DEGs underscores the potential for biological relevance. The model’s high classification accuracy, particularly for PH, reflects the potential utility and promise of these shared genes as diagnostic biomarker candidates. However, the slightly higher classification error observed for IPF patients suggests greater heterogeneity in its genetic and phenotypic presentation. This variability may stem from differences in disease progression, the involvement of secondary pathways, or comorbidities that complicate the underlying molecular landscape. These findings emphasize the need for further refinement of machine learning algorithms and the inclusion of additional multi-omics data to improve diagnostic precision.
Moreover, the correlation patterns among the 13 DEGs provide intriguing insights into the regulatory networks shared by IPF and PH. Strong positive correlations, such as those between PPP1R1A and C14orf180, may reflect coordinated regulatory mechanisms that amplify fibrotic or vascular responses. On the contrary, negative correlations, such as between CNTN6 and PPP1R1A, suggest opposing functional roles in the disease processes, possibly as a result of compensatory mechanisms or divergent signaling pathways. These findings suggest that the interplay between genes involved in fibrosis, immunity, and vascular biology contributes to the pathogenesis of both conditions, offering potential targets for dual-disease therapies. The differential expression patterns between IPF and PH further emphasize distinct molecular mechanisms, highlighting potential targets for developing tailored therapeutic strategies specific to each disease.
It is important to mention that this study points that aminoglutethimide could be a potential drug for the treatment option for IPF associated with PH. The structural and pharmacological profiling of aminoglutethimide highlights its potential as a decent candidate drug for targeting TFF3, a protein implicated in the overlapping molecular mechanisms of IPF and PH. Its bioavailability score of 0.55 and classification under toxicity class 4, with an LD50 of 1800 mg/kg, suggest a favorable safety and efficacy profile. These properties underscore its suitability for further in vitro and in vivo validation in the context of dual-disease pathology. The robust ADMET characteristics, combined with the compound’s interaction score from DGIdb, present aminoglutethimide as a viable candidate in the therapeutic landscape of fibrotic and vascular comorbidities.
In parallel, validation of the TFF3 structural model was performed to ensure reliability in downstream computational analyses. Quality indicators such as 90.2% residues in favored regions of the Ramachandran plot, a PROSA Z-score of −5.62, and favorable G-factor scores confirmed the model’s structural integrity. Furthermore, the amino acid composition analysis revealed a structure rich in stabilizing residues, including cysteine, proline, and alanine. The subsequent molecular docking analysis provided a binding affinity score of −6.1 kcal/mol, indicative of a moderately strong and energetically favorable interaction between aminoglutethimide and TFF3. In clinical practice this drug is used for treatment of seizures, Cushing’s syndrome, breast cancer, and prostate cancer (Santen and Misbin, 1981). However, its potential in other lung disease spectra cannot be overlooked, particularly given its reported efficacy in neutrophil-predominant severe asthma (Xu et al., 2022b). Collectively, these findings provide a strong foundation for future experimental exploration and rational drug design aimed at addressing the shared molecular underpinnings of IPF and PH.
Despite the valuable insights provided, this study has several limitations that warrant consideration. First, the sample size used for the gene expression and machine learning analyses was relatively small, which may limit the generalizability of the findings to broader patient populations. Second, the study relied primarily on transcriptomic data, without incorporating other multi-omics layers such as proteomics or metabolomics, which could provide a more comprehensive understanding of the molecular mechanisms underlying IPF and PH. In addition, while the random forest model demonstrated robust classification performance, the inclusion of more advanced algorithms or ensemble approaches might further enhance predictive accuracy and account for the inherent heterogeneity in these diseases. In addition, model optimization was limited to the use of default parameters. Future studies should consider systematic hyperparameter tuning—such as varying the number of trees and comparing model accuracy across different tree depths—using cross-validation to enhance model generalizability and performance. Another limitation is the lack of experimental validation for the identified genes, as the findings are primarily based on computational analyses. Although validation was performed using an independent dataset, it involved a different biofluid type and focused on CTEPH, which differs biologically from PH associated with LHD. Future validation efforts using datasets specific to PH due to LHD would enhance the robustness and disease relevance of the findings by ensuring alignment within the same subclass of the PH spectrum. Finally, while aminoglutethimide showed promising potential as a therapeutic candidate, its efficacy and safety in the context of IPF and PH need to be rigorously evaluated through preclinical and clinical studies. Addressing these limitations in future research could strengthen the translational relevance of the findings and pave the way for more effective diagnostic and therapeutic strategies.
Conclusions
This study reveals a potential genetic convergence and shared molecular pathways between IPF and PH, highlighting the prospects for unified diagnostic and therapeutic approaches. The consistent dysregulation of 13 genes across both conditions, along with their robust performance in classification models, reinforces their potential as biomarker candidates. Among the therapeutic leads identified, aminoglutethimide emerged as a promising candidate for potential drug repurposing targeting TFF3, a protein that appears to be notably altered in both IPF and PH based on the present in silico study. Its favorable ADMET profile and strong interaction score from DGIdb underscore its therapeutic potential, although further preclinical and in vitro and in vivo studies and validation are necessary and called for. Structural validation and molecular docking analyses further support aminoglutethimide’s potential to effectively bind and modulate TFF3 activity. Despite the limitations of in silico studies, a modest sample size, and reliance on transcriptomic data, the findings offer a valuable framework for future research aimed at precision diagnostics and the development of targeted therapies for patients with IPF and PH. Finally, these findings also challenge the traditional paradigm of pharmaceutical innovation that has long relied on the “one drug, one disease” premise, and highlight the potentials of “one drug, polydisease” paradigm of drug discovery and development.
Footnotes
Acknowledgments
The author is thankful for the permission to use the freely available figure-making tool BioRender.com and the NCBI-GEO tool for the gene expression data.
Author’s Contributions
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing—review/editing.
Author Disclosure Statement
The author declares she has no conflicting financial interests.
Funding Information
No funding was received for the present study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.