
Abstract
The spectrum of childhood interstitial lung diseases (chILD) encompasses a group of heterogeneous, rare disorders in children characterized by diffuse pulmonary infiltrates and disordered gas exchange. Whereas the disorders that present in early life are unique to children, those that present in older children are also seen in adults. This review will concentrate on chILD presenting in children older than 2 years of age with a focus on the idiopathic interstitial pneumonias, connective tissue diseases, alveolar hemorrhage, and hypersensitivity pneumonitis. A systematic approach to diagnosis that includes a careful history and physical, computed tomography of the chest, bronchoalveolar lavage, and lung biopsy can be very helpful in establishing the correct diagnosis. Treatment approaches are described, including general supportive measures, indications for a trial of systemic corticosteroids, or other immunomodulating therapies, and when lung transplantation reserved for those with end-stage lung disease should be considered.
Introduction
As recent attention has been given to the infant with chILD, with the application of a novel classification scheme to diffuse lung disease in young children described by Deutsch et al. 2 ; an Amercian Thoracic Society guidelines for the classification, evaluation, and management of chILD in infants currently near completion; and the review by Deterding in this issue 3 focuses on ILD presenting in children older than 2 years of age. A classification of the disorders seen in this population is presented in Table 1. A thorough discussion of each entity is beyond the scope of this article. Therefore, we will concentrate on the idiopathic interstitial pneumonias, ILD associated with connective tissue diseases, alveolar hemorrhage syndromes, and hypersensitivity pneumonitis. The reader is referred to the reviews by Guillerman 4 and Dishop 5 in this issue for more specific radiologic and pathologic details.
Idiopathic Interstitial Pneumonias
Current classification of the idiopathic interstitial pneumonias in adults is based on a consensus statement of a multidisciplinary panel of pulmonologists, radiologists, and pathologists commissioned by the American Thoracic Society and European Respiratory Society. 6 The panel recognized 7 clinico–radiologic–pathologic entities: idiopathic pulmonary fibrosis, nonspecific interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, desquamative interstitial pneumonia, and lymphoid interstitial pneumonia. With the exception of idiopathic pulmonary fibrosis and respiratory bronchiolitis–interstitial lung disease, the remaining idiopathic interstitial pneumonias are seen in children, although they are quite rare.
Idiopathic pulmonary fibrosis, the most common form of the idiopathic interstitial pneumonia seen in adults, is a progressive and fatal disorder characterized pathologically by usual interstitial pneumonia, a heterogeneous lesion of patchy fibrosis and honeycombing alternating with normal lung and areas of mild to moderate inflammation. The identification of characteristic fibroblastic foci, thought to be the leading edge of the fibrotic process, is required for diagnosis. Although many children have been mistakenly reported as having idiopathic pulmonary fibrosis over the years, the usual interstitial pneumonia pattern with fibroblastic foci has only been described in one adolescent with ABCA3 mutations. 7 Thus true idiopathic pulmonary fibrosis with usual interstitial pneumonia pathology probably does not exist in children.
As in adults, nonspecific interstitial pneumonia in children is considered a distinctive form of ILD that conveys a better prognosis than idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histologically, nonspecific interstitial pneumonia lacks the specific diagnostic features of other idiopathic interstitial pneumonias. It is characterized by a mixture of inflammation and fibrosis and subdivided into a cellular and a fibrosing pattern based on the relative prominence of these particular components. Nonspecific interstitial pneumonia may be seen in a number of clinical settings and with various exposure histories, although most cases are of unknown etiology. It has been described in inborn errors of surfactant metabolism, autoimmune disorders, and hypersensitivity pneumonitis. In adults, the cellular pattern has a better response to steroid therapy and may resolve completely, while the fibrotic pattern is more likely to progress to end-stage lung disease. In a pathologic study of 25 children with ILD, 4 children had cellular nonspecific interstitial pneumonia and 3 had fibrotic nonspecific interstitial pneumonia. 8
Cryptogenic organizing pneumonia, previously called bronchiolitis obliterans organizing pneumonia, has been described in children, either as an isolated phenomenon or in children with infection, asthma, drug reactions, malignancies undergoing chemotherapy, bone marrow transplantation, and autoimmune disorders. 1 As in adults, the prognosis is usually excellent with a favorable response to corticosteroids.
Acute interstitial pneumonia does occur rarely in children with one reported familial case. 9 As in adults, acute interstitial pneumonia in children is a rapidly progressive and often fatal form of idiopathic interstitial pneumonia. The histologic pattern of this clinical entity is diffuse alveolar damage. Corticosteroids and other treatments are often ineffective.
Desquamative interstitial pneumonia has also been reported in children, but in contrast to the adult disorder, it is not related to smoking and carries a worse prognosis. It is now recognized that desquamative interstitial pneumonia in children is almost always caused by an inborn error of surfactant metabolism. 10
Although included as one of the idiopathic interstitial pneumonias, lymphocytic interstitial pneumonia is actually a form of lymphoproliferative disorder, most often seen in patients with immunodeficiency or connective tissue disease. Previously, the most common pulmonary manifestation of pediatric acquired immunodeficiency syndrome, lymphocytic interstitial pneumonia is seen far less frequently today due to the development of improved treatment of human immunodeficiency virus (HIV) infections.
Connective Tissue Diseases
Pulmonary involvement occurs in many types of connective tissue diseases, including systemic lupus erythematosus, systemic scleroderma, dermatomyositis and polymyositis, and Sjogren's syndrome. The general mechanism of injury is the interaction of autoantibodies produced by the body with either the lung parenchyma or the vasculature within the lung resulting in disease.
Systemic lupus erythematosus
The respiratory system is one of the most common organ systems involved in lupus, affecting 80% of adults and 5%–67% of children. 11 Pleuritic chest pain is the most common presenting complaint in children. ILD, occurring in 3%–8% of patients with lupus, is seen less frequently than in other types of connective tissue diseases. Adults are affected more often than children. The most common histologic pattern is nonspecific interstitial pneumonia, but any pattern can be seen.
The most severe and acutely life-threatening pulmonary complication of lupus is acute lupus pneumonitis, a relatively rare disorder with reported incidence of 1%–4% 11 but with mortality rates of 70%–90%. 12 Acute lupus pneumonitis classically presents with acute onset of fever and respiratory distress. Chest films reveal diffuse infiltrates, and lung biopsy when done shows diffuse alveolar damage. It can be difficult to distinguish acute lupus pneumonitis from infectious disease. Interestingly, in half the cases, acute lupus pneumonitis can be the initial presenting manifestation of lupus, adding further difficulty to making the diagnosis. 11 Children can rapidly progress to respiratory failure and death unless treated with corticosteroids.
Systemic sclerosis
In contrast to lupus, ILD is commonly found in systemic sclerosis with up to 25% of adults developing clinically significant disease 12 and with rates of any ILD of 65%–90% in children.13,14 ILD can be seen in both the limited and diffuse types of systemic sclerosis. Although the usual interstitial pneumonia pattern of disease was previously thought to be the most common histologic pattern seen in systemic sclerosis, Desai et al. 15 recently showed that the typical computed tomography (CT) findings in systemic sclerosis included ground glass opacities and reticular opacities resembling nonspecific interstitial pneumonia, in contrast to the predominate pattern of honeycombing and peripheral cysts seen in usual interstitial pneumonia. This has been confirmed in other recent works, and it is now accepted that nonspecific interstitial pneumonia is the most common form of ILD in systemic sclerosis. However, it should be noted that the type of ILD found in these patients has not been correlated to long-term survival. 15
As there have been many studies correlating radiologic and pathologic findings, the diagnosis of ILD in adults with systemic sclerosis is now most commonly made by CT findings alone. As no such pediatric studies exist, lung biopsy may be needed more often in children to confirm the diagnosis. Although not widely available, KL-6 levels have been shown to be significantly elevated in children with systemic sclerosis who had ILD but not in healthy controls nor children with systemic sclerosis without ILD. 16
In a double-blind, placebo-controlled trial in adults with systemic sclerosis, Tashkin et al. 17 demonstrated that daily oral cyclophosphamide therapy resulted in improved forced vital capacity (FVC) and total lung capacity (TLC), less disability, and improved skin thickness score after 12 months of treatment. The same group however found that at 24 months the improvement had waned. 18 A recent consensus group in Europe endorsed cyclophosphamide as the only effective treatment for systemic sclerosis ILD. 19 Cyclophosphamide therapy is cautiously recommended in children with systemic sclerosis and should be given in consultation with a pediatric rheumatologist. Monthly intravenous (IV) cyclophosphamide therapy may offer benefit with fewer side effects than seen with daily oral therapy.
Dermatomyositis/polymyositis
Dermatomyositis/polymyositis are inflammatory myopathies that present with muscle weakness, increased muscular enzymes, changes on electromyography (EMG), and can be associated with ILD. 12 Dermatomyositis includes characteristic dermatologic findings of heliotrope rash and Gotton papules that are absent in polymyositis. Both are associated with the same ILD diseases.
ILD occurs in up to 50% of adults versus 10%–20% of children.20–22 Previously, the most commonly described ILD found in dermatomyositis/polymyositis was usual interstitial pneumonia. 23 However, as in systemic sclerosis, recent reviews now have found that the most commonly described pathologic finding is nonspecific interstitial pneumonia. 12 There also seems to be a larger proportion of cryptogenic organizing pneumonia seen in dermatomyositis/polymyositis than in other connective tissue diseases as well. Cryptogenic organizing pneumonia is often seen in conjunction with nonspecific interstitial pneumonia and is often more treatable than other forms of ILD seen in connective tissue diseases. Although the presence of ILD most often occurs after the diagnosis of dermatomyositis/polymyositis, it can be a presenting sign or precede the diagnosis. 24
The presentation of ILD in the dermatomyositis/polymyositis can be divided into 4 distinct syndromes. 12 The first is the acute presentation that is associated with fever, dyspnea, and diffuse alveolar damage on biopsy and caries a high mortality rate. 11 At times, nonspecific interstitial pneumonia and cryptogenic organizing pneumonia patterns are seen with this presentation, and it is often responsive to treatment. The second pattern is an insidious onset with dyspnea, reduced lung function, and CT findings most often consistent with nonspecific interstitial pneumonia. There is a variable response to therapy. The third presentation is an abnormal CT with no respiratory symptoms, which is usually not clinically significant. The final presentation is abnormal pulmonary function tests and normal CT, which has a variable clinical outcome.
In adults, the treatment of dermatomyositis/polymyositis-associated ILD is steroids and oral or IV cyclophosphamide for severe disease. If the disease is mild, some have used less immunosuppressive regimens such as methotrexate or azathioprine. 11 When present, cryptogenic organizing pneumonia pattern on biopsy or CT is often associated with a positive response to therapy; however, progressive fibrosis can occur. 24 In a report by Kobayashi et al., 22 a group of 3 children with ILD and dermatomyositis were treated with cyclosporine A after failing prednisone alone and 2 showed improvement.
Sjogren's syndrome
Sjogren's syndrome is a disease characterized by the triad of xerostomia, xerophthalmia, and arthritis, and lymphocytic infiltration of various organs in the body, including the airways and lungs. 11 Sjogren's syndrome presents as either primary Sjogren's syndrome or secondary Sjogren's syndrome that is associated with other connective tissue diseases including rheumatoid arthritis and lupus. The prevalence of ILD in Sjogren's syndrome varies from 9% to 75% of patients.
ILD is more commonly found in patients with secondary Sjogren's syndrome than primary Sjogren's syndrome. While nonspecific interstitial pneumonia is the most common pattern of disease in Sjogren's, there is more pattern variability compared with other connective tissue diseases. Lymphocytic interstitial pneumonia is particularly more prevalent in Sjogren's syndrome than in other collagen vascular diseases. 12
There is no consensus on treatment of Sjogren's syndrome. Corticosteroids seem to be effective in preventing progression and at times causing regression of the disease. Deheinzelin et al. 25 studied a small group of patients who were either treated with azathioprine with and without steroids or placebo. The treatment group had more improvement in lung disease versus the placebo group, suggesting that azathioprine may be effective in the treatment of Sjogren's syndrome.
Alveolar Hemorrhage Syndromes
Alveolar hemorrhage occurs when the small pulmonary vessels in the lung are damaged, allowing blood to leak into the alveolar spaces. 26 While the presentation is often acute with hypoxemia, cough, dyspnea, alveolar infiltrates on chest films, and hemoptysis, it can also present insidiously with anemia, cough, and exercise limitation.26–29 Hemoptysis may not been seen in young children as they tend to swallow their secretions. As shown in Table 2, alveolar hemorrhage can be classified into 2 groups, those with and those without pulmonary capillaritis. The group of alveolar hemorrhage disorders without pulmonary capillaritis can be further subdivided into those with and without cardiac disease. Idiopathic pulmonary hemosiderosis would be considered in the latter category.
Pulmonary capillaritis
Pulmonary capillaritis is a small vessel vasculitis with features of neutrophilic invasion followed by fibrinoid necrosis of the capillary walls. 30 It can be a primary, isolated disorder or associated with other more specific vasculitides, such as Wegener's granulomatosis, microscopic polyangiitis, and Goodpasture's disease. Pulmonary capillaritis is an underrecognized cause of alveolar hemorrhage and is often misdiagnosed as idiopathic pulmonary hemosiderosis. In a series of 23 children with pulmonary hemorrhage, Fullmer et al. 30 found pulmonary capillaritis in 8 and idiopathic pulmonary hemosiderosis in 4.
Pulmonary capillaritis is often but not always associated with anti-neutrophilic cytoplasmic antibodies (ANCA). Classically, anti-proteinase 3 antibodies (c-ANCA) are found in Wegener's granulomatosis, while anti-myeloperoxidase antibodies (p-ANCA) are found in microscopic polyangiitis. 29 Goodpasture's disease is associated with antiglomerular basement membrane antibodies. 27 These 3 disorders often involve the kidneys as well and thus are known as pulmonary–renal syndromes. In isolated, primary pulmonary capillaritis, autoantibodies may not be detected in serum. In secondary pulmonary capillaritis, the characteristic histologic lung findings can precede renal involvement and positive serum markers.26,29 In these cases, lung biopsy is essential in differentiating pulmonary capillaritis and idiopathic pulmonary hemosiderosis. Fullmer et al. demonstrated that children with pulmonary capillaritis had a significantly high erythrocyte sedimentation rate at baseline compared with idiopathic pulmonary hemosiderosis. 30
Aggressive treatment is often required as pulmonary capillaritis is often associated with high morbidity and mortality. In our experience, corticosteroids alone are seldom effective in controlling disease activity, and additional therapy is often required. The goals of treatment are first to induce and then to maintain remission. Induction therapy for most patients includes high-dose corticosteroids (given either by mouth daily or by IV pulse, once weekly or 3 consecutive days monthly) and cyclophosphamide and/or IV immunoglobulin. Maintenance therapy includes low-dose prednisone and either azathiaprine or methotrexate. A randomized, nonblinded study by Pagnoux et al. 31 of 126 adult patients with Wegener's granulomatosis or microscopic polyangiitis found no difference between azathiaprine or methotrexate; however, no comparable study has been done in children. Although older studies suggested a poor prognosis with mean survival of under 3 years, recent studies are associated with an improved survival of greater than 80%. 26
Idiopathic pulmonary hemosiderosis
Idiopathic pulmonary hemosiderosis is defined by the classic triad of anemia, infiltrates on CXR and hemoptysis, although, as in other causes of alveolar hemorrhage, the hemoptysis is not always present in young children. 27 It is a diagnosis of exclusion and is often made in the literature with the combination of the above and the presence of hemosiderin-laden macrophages in bronchoalveolar lavage (BAL) or gastric aspirates. 32 The onset of idiopathic pulmonary hemosiderosis can be acute and life-threatening, sometimes requiring intubation and mechanical ventilation. 33
Some authors have argued that the above triad combined with negative autoantibody titers to the diseases associated with pulmonary capillaritis is sufficient to make the diagnosis of idiopathic pulmonary hemosiderosis and that lung biopsy is rarely indicated.32,33 However, in the pathologic review of 23 children with pulmonary hemorrhage, only 4 were diagnosed with idiopathic pulmonary hemosiderosis versus 8 with pulmonary capillaritis, suggesting that pulmonary capillaritis is much more common than idiopathic pulmonary hemosiderosis. 25 Since some children with pulmonary capillaritis have negative serology, the only reliable way to differentiate the 2 diseases is with a lung biopsy. This biopsy should be reviewed by an experienced pediatric pulmonary pathologist, as the pathologic features of pulmonary capillaritis can be subtle. Since idiopathic pulmonary hemosiderosis is a diagnosis of exclusion, serology of some patients with pulmonary capillaritis is negative, and most patients with idiopathic pulmonary hemosiderosis do not have a tissue diagnosis, it is likely that some patients with idiopathic pulmonary hemosiderosis actually have pulmonary capillaritis.
This distinction is important because most children with idiopathic pulmonary hemosiderosis can be managed with corticosteroids alone in contrast to those with pulmonary capillaritis who may require more aggressive therapy. Since most of the children in idiopathic pulmonary hemosiderosis studies did not have a lung biopsy, it is possible that some of these patients who seemed to respond to steroid-sparing agents such as 6-mercaptopurine actually had pulmonary capillaritis. 30
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis, results from an abnormal immune response to inhaled organic particles. Although more common in adults, hypersensitivity pneumonitis is seen in children mainly older than 2 years of age. In a review of pediatric hypersensitivity pneumonitis, the average age at diagnosis was 10 years old. In children with hypersensitivity pneumonitis, the most common etiology is bird antigens.34–37 It can also result from exposure to a wide variety of organic antigens such as molds such as from moldy hay or grains, aerosolized molds, and mold contaminated living areas. Highly reactive low-molecular-weight chemicals such as in spray paints, some glues, epoxy resins, and some insecticides can also cause hypersensitivity pneumonitis. There are case reports implicating other organic antigens, including cat, 38 as potential causes of hypersensitivity pneumonitis. A number of drugs can cause a hypersensitivity pneumonitis-like picture; a searchable database of drug-induced lung diseases is available at www.pneumotox.com. It is particularly important to note that methotrexate can cause hypersensitivity type ILD, 34 as this medication is often used in treating connective tissue diseases.
The pathogenesis of hypersensitivity pneumonitis is not completely understood. An exuberant production of IgG antibody against the offending antigen is usually identified, but is not causative. 39 Cell-mediated reactions, including the cells like T lymphocytes and natural killer T cells, 40 are believed to play an important role.34,36,41 Various cytokines are elevated in hypersensitivity pneumonitides, including IL-8, IL-12, TNF-α, and interferon γ.42–44 It seems that the initial exposure causes an increase in IgG, which results in immune complex deposition. These immune complexes cause the recruitment of macrophages to the lungs. The macrophages produce a number of chemokines including IL-8, macrophage inflammatory protein α1, and RANTES, which recruit T cells.34,41 T cells then produce other chemokines such as interferon γ, which leads to granuloma formation within the lung. 34 As the disease progresses, other chemokines are produced. This leads to a general increase in T cells in the lungs with a large increase in CD8+ T cells.
As in adults, hypersensitivity pneumonitis in children can occur in 3 forms: acute, subacute, or chronic. In the acute form, fever, cough, and body aches develops 4–8 h after exposure to the antigen. This presentation is often confused with an acute viral illness or acute infectious pneumonia, but it can be differentiated by its repeated occurrence after antigen exposure. Acute HP can cause severe, life-threatening reactions. In the subacute form, 34 children gradually develop chronic cough, weight loss, low grade fever, hypoxemia, and crackles on auscultation. They often have diffuse reticulonodular infiltrates on chest films and ill-defined micronodules on CT scan. Chronic hypersensitivity pneumonitis has an insidious onset and is often difficult to diagnose in its early stages. Over time chronic, progressive symptoms develop with chronic cough, dyspnea, fatigue, weight loss, and exercise intolerance. In its late stages radiologic findings of lung damage due to pulmonary fibrosis, emphysema, and honeycombing can be seen. 45
Major and minor criteria have been developed for the diagnosis of hypersensitivity pneumonitis in adults. Major criteria include: (1) symptoms compatible with hypersensitivity pneumonitis, (2) evidence of exposure to the proper antigen by history or by detection of antibodies in the serum or BAL, (3) findings on CT or chest X-ray compatible with hypersensitivity pneumonitis, (4) BAL lymphocytosis if BAL was performed, (5) pulmonary histologic changes compatible with hypersensitivity pneumonitis if lung biopsy was performed, (6) positive natural challenge such as antigen challenge or reexposure to the antigen causing similar symptoms. Minor criteria include: (1) bibasilar crackles, (2) decreased diffusing capacity, and (3) arterial hypoxemia. A confident diagnosis can be made when a patient has 4 major and 2 minor criteria.34,36 Serum precipitins are not always reliable in patients with hypersensitivity pneumonitis. In a study of 121 adults in which hypersensitivity pneumonitis was a consideration, Fenoglio et al. 46 reported that precipitins had a positive predictive value of 71%–75% with a negative predictive value of 81%–88% in the 31 patients with confirmed hypersensitivity pneumonitis. An expert panel review of hypersensitivity pneumonitis also concluded that precipitins, while important, are not sufficiently sensitive. There are reports of initially false negative precipitin studies. 47 A lack of positive precipitins should not exclude the diagnosis. 41 Positive precipitins can also be found in exposed individuals without disease. 48
The use of these criteria in children has some limitations. While BAL lymphocytosis and a low CD4+/CD8+ ratio occur in both children and adults with hypersensitivity pneumonitis, 49 healthy children also have a low CD4+/CD8+ ratio, in contrast to healthy adults who have a ratio of 1.50,51 Thus the CD4+/CD8+ ratio is not useful for the diagnosis of hypersensitivity pneumonitis in children. In addition, diffusion lung capacity for carbon monoxide (DLCO) cannot be reliably performed in young children. Thus we recommend that the diagnosis in children be based on compatible symptoms and physical findings, history of exposure and/or positive precipitins, diagnostic imaging studies consistent with the diagnosis, BAL lymphocytosis, if BAL performed, characteristic histologic features if biopsy performed, and clinical improvement with antigen elimination.
The primary treatment is eliminating the patient's exposure to the antigen, which is often curative. The use of corticosteroids, given orally daily or in monthly IV pulses, is usually effective in hastening recovery. The prognosis in children is excellent, especially if the disease is discovered before permanent lung damage has occurred and the offending antigen is identified and exposure eliminated. Rarely, symptoms may progress despite apparent antigen removal and systemic corticosteroids. In contrast to adults, mortality from pediatric hypersensitivity pneumonitis is rare 52 ; however, severe disease and life-threatening complications have been reported.
Approach to Diagnosis
As the differential diagnosis is extensive, a systematic approach is essential for accurate diagnosis. A thorough history that reviews environmental exposures, respiratory symptoms related to feeding, other organ involvement, a history of severe respiratory infections, including adenovirus, and the presence of hemoptysis should be obtained. A physical exam should look for the presence of growth failure, tachypnea, crackles, loud P2, and clubbing.
Pulmonary function testing, 6-min walk test, and pulse oximetry may be used to assess severity of disease. High-resolution computed tomography (HRCT) may detect extent and severity of disease. The HRCT findings may suggest a specific diagnosis, such as bronchiolitis obliterans (mosaic perfusion, vascular attenuation, and central bronchiectasis), hypersensitivity pneumonitis (ill-defined centralobular micronodules), and pulmonary alveolar proteinosis (crazy-paving). Bronchoalveolar lavage can show evidence of alveolar hemorrhage syndromes (hemosiderin-laden macrophages), aspiration (lipid-laden macrophages), hypersensitivity pneumonitis and sarcoidosis (lymphocytosis), eosinophilic pneumonias (eosinophilia), and histiocytosis (CD1a+ cells). Although lung biopsy remains the gold standard for diagnosis of chILD, it must be interpreted in the context of the clinical and radiologic findings. Figure 1 shows a simplified algorithm for the diagnosis of chILD presenting in children older than 2 years of age. It should be emphasized that although lung biopsy may not be necessary when less invasive studies such as HRCT are able to establish the diagnosis.
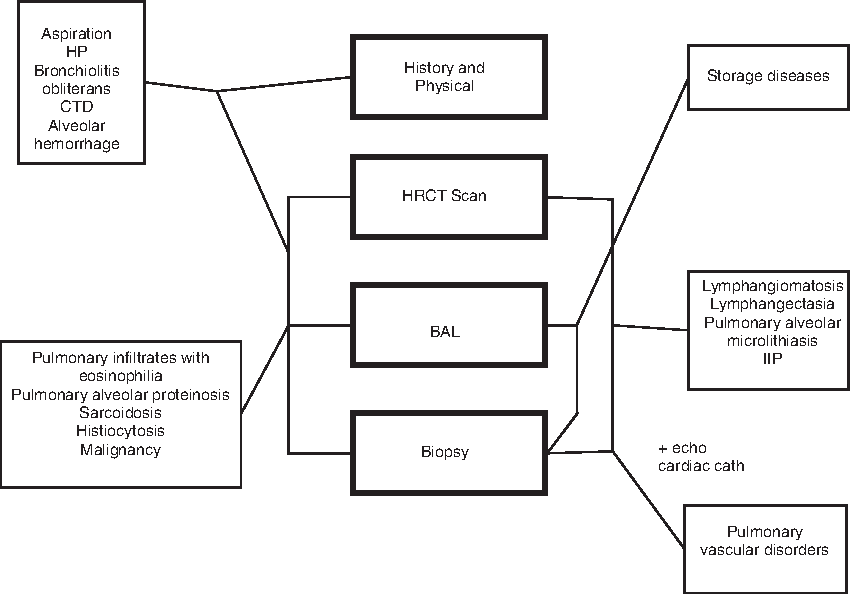
Algorithm for the diagnosis of interstitial lung disease (ILD) presenting after 2 years of age. The connecting lines indicate which procedures are helpful in diagnosing each disorder. For example, lymphangiomatosis can be diagnosed with a high-resolution computed tomography (HRCT) and biopsy alone. Not all procedures are necessary for every diagnosis; for example, hypersensitivity pneumonitis (HP) often requires a history and physical, HRCT scan, and a bronchoalveolar lavage (BAL), but not a biopsy. CTD, connective tissue disease; IIP, idiopathic interstitial pneumonia.
Treatment
Supportive care, generalized to all patients with chILD, includes aggressive nutritional intervention, prevention of infection through influenza and other vaccines, supervised fitness and pulmonary rehabilitation programs, avoidance of inhaled tobacco smoke and other irritants, and family support. Voluntary health organizations, such as the chILD Foundation (www.childfoundation.us), can provide important support and education resources. As in cystic fibrosis, a multidisciplinary team approach that includes the patient's primary care physician, pulmonologist, nutritionist, social worker, respiratory nurse, and respiratory therapist is highly desirable to meet the needs of these families whose children often have complex medical issues.
Many children with advanced disease require supplemental oxygen either at night or continuously. The development of pulmonary hypertension is associated with a decreased probability of survival in chILD. 53 Thus, patients with pulmonary hypertension should receive oxygen and pulmonary vasodilator therapy, such as sildenafil or bosanten, should be considered.
As inflammation is thought to play an important role in many forms of chILD, corticosteroids with their multifaceted anti-inflammatory properties are considered first-line therapy for nonspecific interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia, desquamative interstitial pneumonia, lymphocytic interstitial pneumonia, hypersensitivity pneumonia, eosinophilic pneumonia, alveolar hemorrhage, and many connective tissue diseases with pulmonary involvement. However, therapy is empiric as no prospective, controlled clinical trials exist for any of these disorders in children. Corticosteroids can be given in oral, daily doses of prednisone or prednisolone, 1–2 mg/kg/day or in IV pulse doses of methylprednisolone, 30 mg/kg, with a maximum of 1 g, given once weekly or on 3 consecutive days monthly. While no comparative trials exist, the authors' experience is that pulse steroid therapy is associated with fewer side effects. Steroid-sparing agents, such as hydroxychloroquine, azathioprine, methotrexate, cyclophosphamide, tacrolimus, meclofenamate mofetil, and immunodulatory doses of IV immunoglobulin, have been used in children with varying success, again with no clinical trials of their efficacy.
For some diseases, there are more specific therapies. For hypersensitivity pneumonitis, antigen removal is critical. For pulmonary alveolar proteinosis caused by autoantibodies to granulocyte macrophage colony-stimulating factor (GM-CSF), treatment is inhaled GM-CSF plus repeated whole lung lavage to remove the proteinaceous material is often quite effective in controlling the disease process.54,55 Finally, for pulmonary hemangiomatosis, treatment with interferon α can result in regression of the vascular proliferation. 56
Lung transplantation is an option for chILD that progresses to end-stage lung disease. Our experience is that outcomes for transplantation for chILD are comparable with that for transplantation for cystic fibrosis or pulmonary hypertension.
Summary
chILD presenting after 2 years of age is a heterogeneous group of disorders that overlap with ILD seen in adults, but differs considerably from ILD presenting in infancy. Some are primary lung disorders while others are associated with systemic disease processes. A systematic approach that includes a thorough history and physical exam, and selected use of HRCT, BAL, and lung biopsy is valuable for accurate diagnosis. Although the evidence is strictly experiential, systemic corticosteroids are commonly used as first-line therapy for most of these diseases with steroid-sparing agents required in some cases. Due to the relative rarity of these diseases, multicenter collaboration through groups like the ChILD Research Network (CHILDRN) is essential for study of these challenging and diverse diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.