
Abstract
Pulmonary alveolar microlithiasis is rare in children. It presents as a chronic disease with early asymptomatic course and is usually idiopathic. Characteristic radiology and high index of suspicion can lead to early diagnosis by lung biopsy. Treatment with drugs targeting phosphate metabolism may be beneficial. We present a series of 3 pediatric cases confirmed by lung biopsies, along with review of literature.
Introduction
Case 1
A 7-year-old boy presented with nonproductive cough for 3 years with dyspnoea on exertion for the preceding 1 year. He was otherwise healthy. There was no history of consanguinity or contact with tuberculosis. On examination, height was 120 cm and weight 20 kg (both at 50th centiles for age). General and systemic examination did not reveal any abnormality. Resting oxygen saturation was 100% on room air. Baseline hematological investigations were normal. Chest radiograph showed bilateral micronodular opacities obliterating the cardiac, mediastinal, diaphragmatic borders, and the interlobar fissures. They were concentrated mainly over the lower two-thirds of both the lung zones (Fig. 1). Mantoux skin test was nonreactive. Three gastric lavages for acid-fast bacilli stain and culture were negative. Pulmonary function tests (PFT) revealed a mild restrictive pattern. High-resolution computed tomography (HRCT) showed miliary opacities located in the same distribution as described above (Fig. 2). Based on the clinical picture and radiography, a provisional diagnosis of PAM was made. Transbronchial lung biopsy (TBLB) and histopathological examination confirmed the presence of alveolar microliths (Fig. 3). Family screening by chest radiography was negative.
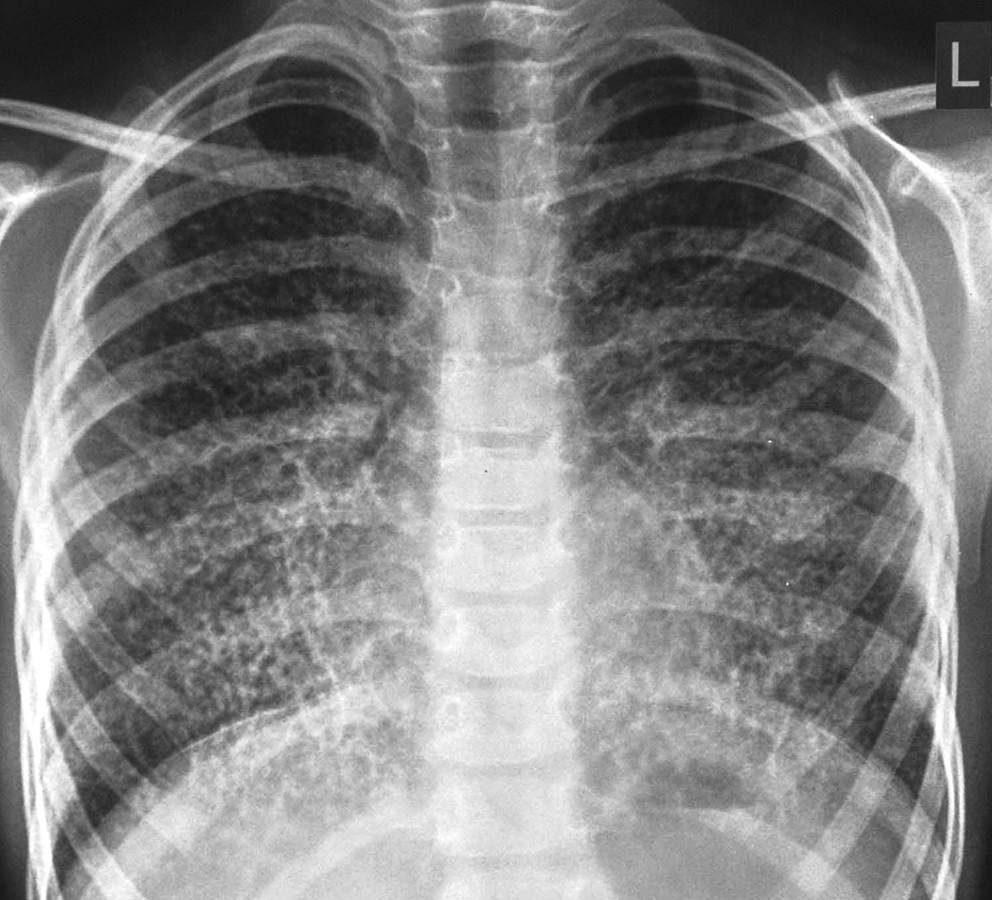
Chest radiograph showing bilateral micronodular and reticular pulmonary opacities bilaterally, most prominent at the lower lung zones and partially obscuring the cardiac and diaphragmatic margins.
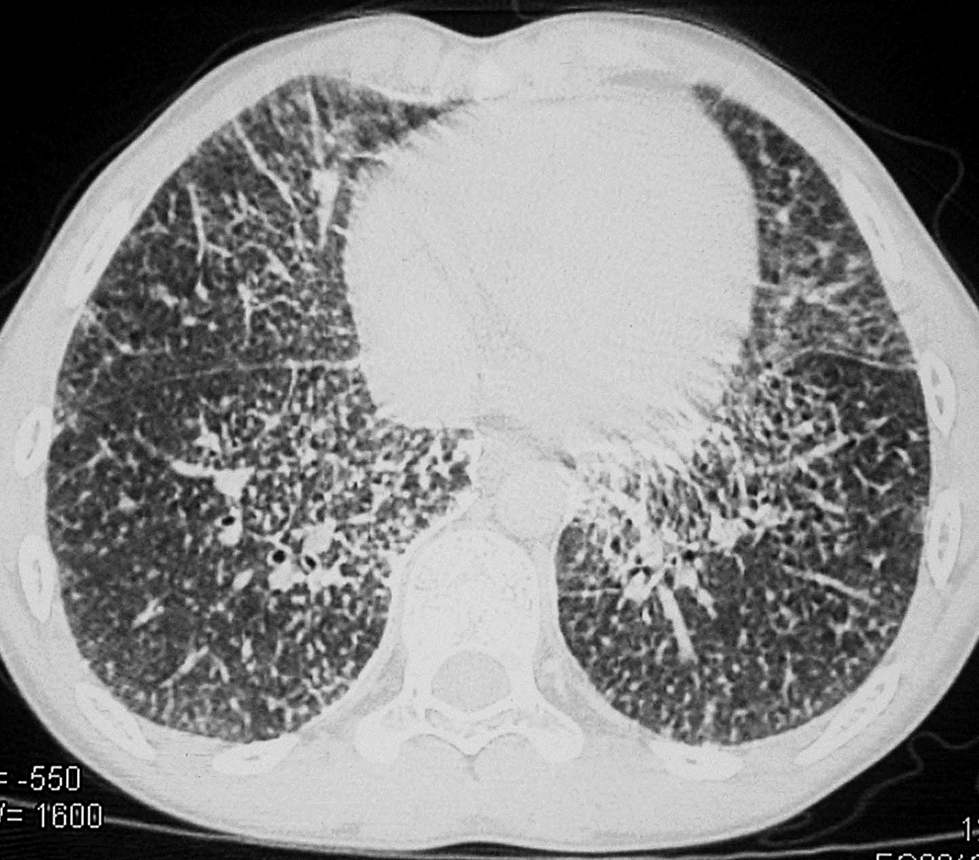
Chest high-resolution computed tomography image showing a micronodular and reticular pattern.
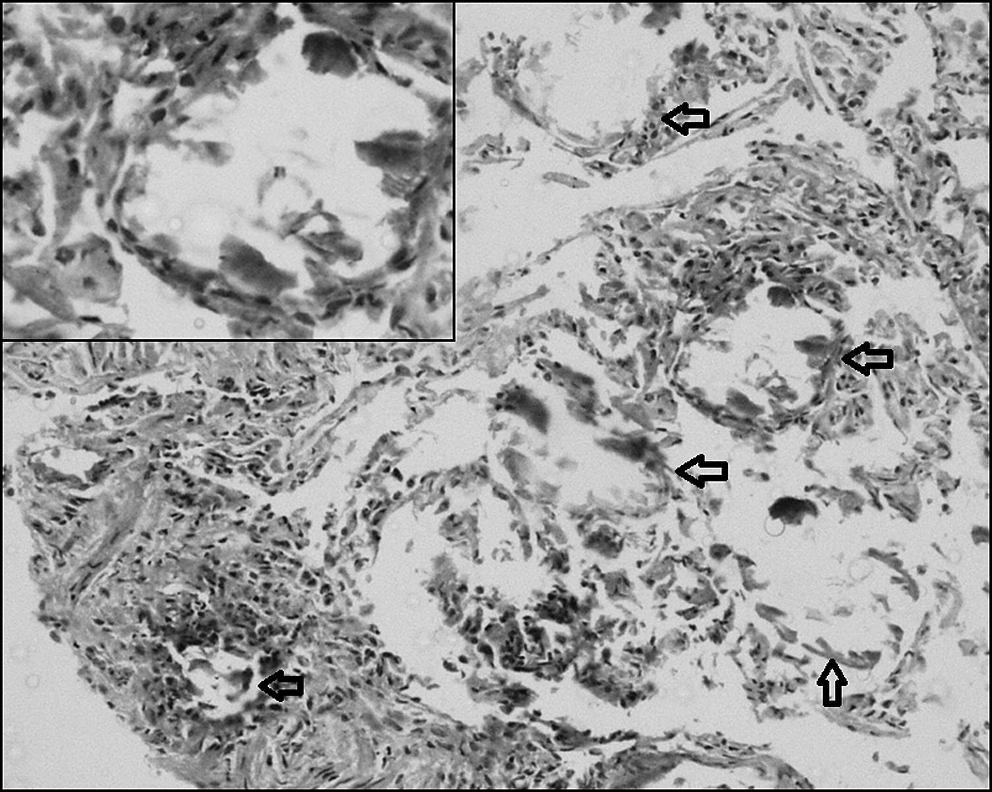
Photomicrograph of lung biopsy specimen showing few alveolar spaces (arrows) with calcifications and formation of microliths (hematoxylin and eosin, ×280). Inset shows higher magnification.
Case 2
A 12-year-old boy presented with intermittent cough for 7 years with production of white, mucoid sputum. There was no history of fever, weight loss, or hemoptysis. He had received antituberculous therapy for 9 months 3 years before but symptoms did not remit. There was no history of contact with tuberculosis. On examination, height was 150 cm and weight 38 kg (both at 50th centiles for the age). Rest of the systemic examination was unremarkable. Chest radiograph was suggestive of bilateral diffuse reticulonodular lung opacities. Mantoux test was nonreactive. Three sputum samples for acid-fast bacilli stain and culture were negative. PFT showed a mild restrictive pattern. HRCT revealed multiple subcentimetric discrete homogenous lymph nodes in the precarinal, subcarinal, and pretracheal regions with interlobar septal thickening and multiple, small (<5 mm) perilymphatic nodules in both the lung zones. Sarcoidosis was initially considered but angiotensin-converting enzyme level was normal. TBLB was done; histopathology confirmed the presence of alveolar microliths. Broncho-alveolar lavage fluid staining for calcium with Papanicolou and Von Kossa stains was negative. Family members did not have abnormal radiographs.
Case 3
A 6-year-old girl was admitted with chronic cough and low grade, intermittent fever for 6 months. There was no history of contact with tuberculosis. On examination, weight was 17 kg and height 110 cm (both at 50th centiles for age). General and systemic examination was normal. Chest radiograph was suggestive of miliary opacities over both the lung zones. Mantoux skin test was reactive (10×12 mm) but 3 gastric lavages for acid-fast bacilli stain and culture were negative. HRCT showed similar miliary mottling over both the lung zones. She was initially treated with antituberculous therapy for 6 months following which the fever and general condition improved but mild cough persisted; there was no change in the chest radiograph in follow-up. Hence, a provisional diagnosis of PAM was considered for which TBLB was done which revealed microliths. Family screening for PAM by chest radiography was negative.
Discussion
PAM is a rare, chronic pulmonary disease characterized by deposition of lamellar concretions of calcium phosphate within the alveoli, creating a classic “sandstorm” pattern on the chest radiograph and a progressive restriction of lung functions. The first macroscopic description of the disease was given by Malpighi in 1966. 2 Puhr coined the term “Pulmonary alveolar microlithiasis” in 1933. 3 The condition being uncommon in children is often confused with other causes of miliary lung opacities like miliary tuberculosis, fungal infections, metastases, varicella pneumonia, pulmonary alveolar hemosiderosis, sarcoidosis, and pulmonary adenomatosis. 4 They often receive antituberculous therapy and are investigated for sarcoidosis and malignancy.5,6 Hence a high index of suspicion and acquaintance with characteristic imaging findings is required.
The etiology is postulated to be genetic. The former view was an inherited metabolic abnormality of carbonic anhydrase enzyme, which promotes alkalinity of the alveolar surface, with consequent precipitation of calcareous salts. 7 Recently, the gene SLC34A2 related to the disease has been discovered which encodes a type IIb sodium phosphate co-transporter in the type 2 alveolar cells in the lungs. The function of the gene protein is to uptake liberated phosphate from alveolar fluid for the phospholipid component of the surfactant, produced by the type 2 alveolar cells. A loss-of-function mutation of SLC34A2 is postulated to reduce the clearance of phosphate, thus leading to the formation of microliths by calcium-phosphate chelation. 8 The mutation analysis could not be done in any of our 3 patients due to financial constraints.
Clinical presentation has been described as early as the newborn period, 9 including premature babies. 10 Most affected children are initially asymptomatic with clinico-radiological dissociation. Later they develop chronic cough, dyspnoea on exertion, hemoptysis, digital clubbing, hypertrophic pulmonary osteoarthropathy, 11 or recurrent pneumothoraces. Ritchie et al. 12 incidentally discovered PAM in a 5-year-old boy being evaluated for multiple exostoses. PAM with calcified pericardium 13 and gallstones 6 is also described. Autosomal recessive mode of inheritance has been suggested in familial cases. 14 Progressive diffuse interstitial fibrosis, alveolar wall destruction, diminished vascularity, pulmonary hypertension, and cor pulmonale characterize the end-stage disease. 15 Asymptomatic children with PAM have been diagnosed by family screening of adult patients, reiterating the importance of chest radiography in family members.16,17 The familial form of the disease has a genetic origin and there is poor correlation between genotype and phenotype which forms an important part of counseling. In all 3 of our patients, the family members had normal radiographs.
Chest radiography is characteristic and is the most important clue to the diagnosis. HRCT chest findings of ground glass attenuation with superimposed septal thickening and calcifications along the interlobar septa have been described as diagnostic of PAM.16,18 The distribution of the calcific nodules at the lung bases can be explained by the relative higher blood supply to this area. Additionally, small apical bullae; black pleural line seen as an area of increased translucency between the lung parenchyma and ribs; or pleural thickening seen due to heavy concentration of microliths in the subpleural parenchyma may be appreciated. Pulmonary functions gradually show restriction of lung volumes and impairment of gas exchange with disease progression. Tc99m-diphosphonates bone scintigraphy 19 and staining of BAL fluid with Von Kossa and Papanicolou stains 20 has also been used for diagnosis but histopathological demonstration of microliths by lung biopsy is confirmatory.
Specific treatment options are still at the experimental stage; steroids have not been helpful. In advanced restrictive disease, lung transplantation is the only effective treatment. It is not known whether PAM recurs after transplantation. Recurrence after transplantation has not been reported till date. 21 Genetic studies show a future potential for drugs targeting phosphate metabolism rather than the calcium metabolism. Due to their inhibitory effect on the formation of calcium phosphate crystals in the body, bisphosphonates have been used by Ozcelik et al. 22 in 2 patients of PAM. Long term monitoring for obvious side effects of bisphosphonates therapy like rickets, hyperphosphatemia, and pathological fractures is required. No bisphosphonate therapy was administered to our patients as it may be deleterious to the growing skeleton and larger studies are needed before its widespread use. They have remained well in follow-up for 5 years till now with no worsening of pulmonary functions. The follow-up chest radiographs have not shown any change.
Conclusion
PAM is characterized by chronic respiratory symptoms and characteristic radiology. It is often misdiagnosed as miliary tuberculosis; hence alternative diagnoses must be considered if other tests are negative and/or response to antituberculous therapy is unsatisfactory. TBLB and histopathological examination confirm the diagnosis. Screening of the family members with radiography is recommended. There are limited treatment options at present.
Footnotes
Author Disclosure Statement
No competing financial interests exist.