
Abstract
Data from the New Drug Application for inhaled dry powder mannitol (DPM) use in cystic fibrosis in children and adults ages 6 years and older was reviewed and discussed at the Food and Drug Administration Pulmonary-Allergy Drugs Advisory Committee meeting in January 2013. Of the two pivotal Phase III trials (CF-301 and CF-302), only one demonstrated efficacy for the primary endpoint—absolute change from baseline Forced Expiratory Volume in 1 second (FEV1 mL). In addition, neither trial demonstrated efficacy in the pediatric population who comprised 43% of the intent-to-treat study population. Also, there was an increased risk of hemoptysis in children receiving DPM versus control, and the difference in rates between DPM and control was larger than that observed in adults. This review will describe the main issues discussed for the pediatric population. All study data presented in this review are publically available from the Food and Drug Administration (FDA) Web site at www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Pulmonary-AllergyDrugsAdvisoryCommittee/ucm329187.htm for the January 30, 2013 meeting.
I
Mannitol is a nonionic sugar alcohol that is not readily metabolized, nor transported across airway epithelium, and is poorly absorbed. 1 As a hyperosmotic agent, its beneficial feature in CF is to promote influx of fluid into the periciliary liquid layer and improve mucociliary clearance by increasing the volume of the periciliary liquid layer. 2 The only other osmotic agent currently used for this purpose is nebulized hypertonic saline, but it is not FDA approved for this indication in patients with CF. DPM induces changes in the properties of sputum, including reducing the percent of solids present and decreasing surface tension. 1 Changes in viscoelasticity were not seen in a 2 week study in CF in contrast to findings in patients with asthma or bronchiectasis, presumably because of the larger variability in sputum viscoelasticity in CF due to the greater proportion of pus.1,3 The beneficial effect of DPM is likely related to promoting a more effective cough.
The PADAC was asked to discuss the efficacy of DPM 400 mg twice daily in improving lung function and the safety data separately in adults and children. However, the PADAC was required to vote on the efficacy and safety data in the total population of adults and children and was not allowed to alter the voting questions, though some members would have preferred a separate vote for children and adults. The FDA was not bound by the vote of the PADAC in its decision. The Federal Food, Drug, and Cosmetic Act (21 Code of Federal Regulations Chapter 9 Part A Sec 355) 4 requires “substantial evidence” consisting of “adequate and well-controlled investigations” (21 Code of Federal Regulations Chapter 1 Food and Drug Administration, Department of Health and Human Services, Subchapter D, Part 314, Subpart D, Section 314.126) 5 that a drug will have the effects claimed in order to be considered for market approval. The FDA has interpreted this to mean there must be “at least two adequate and well-controlled Phase III clinical studies, each providing convincing evidence of effectiveness.” 6
The data presented to the PADAC were from two Phase III trials, referred to as CF-301 and CF-302, that tested a 400 mg dose of DPM twice daily (10 capsules of 40 mg inhaled separately) in a breath actuated inhaler (Dry Powder RS01 Inhaler Model 7; Plastiape, Milan, Italy). These trials were conducted consecutively, and the study design was nearly identical for both trials, consisting of a randomized double-blind controlled 26 week study of 400 mg versus 50 mg (control) DPM twice daily followed by a 52 week and 26 week open label phase (CF-301 and CF-302 respectively) in which all participants received 400 mg DPM twice daily. The 50 mg DPM dose was found not to have any beneficial or adverse effects in earlier clinical testing, and was used as a control to mask the sweet taste of mannitol. The inclusion and exclusion criteria were similar, except in CF-302, participants were required to have a FEV1 of at least 40% of predicted, whereas in CF-301, which was the first trial conducted, participants could have a baseline FEV1 of 30% of predicted. Importantly, patients with a history of significant hemoptysis (>60 mL within 3 months of enrollment) were excluded. Because inhaled mannitol is known to cause bronchospasm, eligible participants were given a mannitol bronchoprovocation test (maximum dose 395 or 400 mg in CF-301 and CF-302 respectively) to screen out participants with airway hyperresponsiveness. The primary efficacy endpoint for both trials was the absolute change from baseline FEV1 (mL). Although DPM is not a bronchodilator, this endpoint was considered appropriate as pulmonary function progressively declines by several percent per year in patients with CF so any improvements would be considered clinically meaningful by the FDA. Although an endpoint using exacerbation rates would be preferable in this population, the larger sample size and longer duration of study that would have been required precluded this approach.
The initial concern of the PADAC was that each trial, CF-301 and CF-302, did not demonstrate statistically significant improvements in the absolute change from baseline FEV1. In CF-301 and CF-302, the difference from control was 83.1 mL [95% CI 39.5, 126.8] (p<0.001) and 54 mL [95% CI −2.0, 110.3] (p=0.059) respectively. Thus, the trials did not meet the FDA requirement for two trials demonstrating efficacy for the proposed claimed effects. Under certain circumstances, the FDA may use data from one adequate and well-controlled trial, for example, if the secondary endpoints are strongly supportive and consistent with the primary endpoint. However, the secondary nonpulmonary function endpoints, which included exacerbation rate, rescue antibiotic use, quality of life questionnaires, and hospitalizations for exacerbations, were also not significant. In the combined CF-301 and CF-302 trial analysis, children (6–11 years) and adolescents (12–17 years) comprised approximately 43% of the intent-to-treat population. The improvement in absolute FEV1 in children and adolescents in the combined analysis of CF-301 and CF-302 was 20.1 mL [95% CI −52.7 to 92.9] and 73.7 mL [−14.3 to 161.7] respectively. Thus, efficacy was not demonstrated in children, and the lower limit of the confidence intervals indicates that the true mean could actually be a decrease in FEV1 from baseline. Because of the high dropout rate (described below), responder analyses (improvement in FEV1 of 100 mL) were generated by the FDA that showed a differential response in pediatric participants compared to the intent-to-treat population as a whole for each study. In pediatric participants in CF-301, little or no difference was noted in the responder analysis between DPM and control, whereas in CF-302, some separation is seen (Fig. 1). However, the odds ratio for an absolute increase in FEV1 of ≥100 mL in CF-301 in children aged 6–11 years was 1.09 [95% CI 0.26, 4.48] (p=0.908), and in adolescents aged 12–17 years it was 0.86 [0.27, 2.73] (p=0.803). In CF-302, the odds ratios were 2.25 [95% CI 0.66, 7.72] (p=0.196) in children aged 6–11 years and 1.25 [95% CI 0.48, 3.30] (p=0.639) in adolescents. Thus, these results indicate no statistically significant improvement in pediatric patients treated with DPM on FEV1.
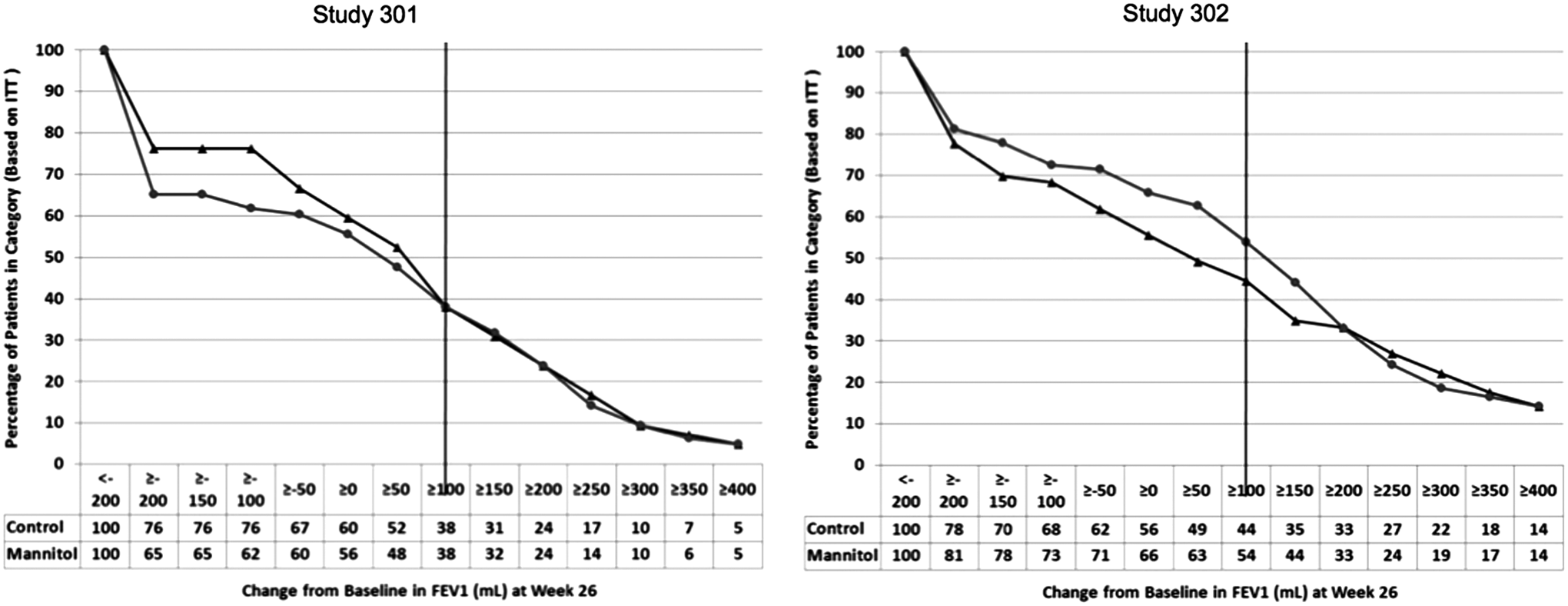
Post hoc responder analysis of change in baseline forced expiratory volume in 1 second (FEV1) (mL) at week 26 in pediatric participants aged 6–17 years in studies CF-301 and CF-302. The vertical line represents those pediatric participants who achieved a threshold 100 mL change. The initial drop from 100% on the y-axis corresponds to proportion of participants who discontinued the study before week 6 and are considered missing data. Control is shown by triangles; DPM is shown by circles. Data presented by the FDA at the PADAC Meeting January 30, 2013. www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Pulmonary-AllergyDrugsAdvisoryCommittee/ucm329187.htm. DPM, dry powder mannitol; FDA, Food and Drug Administration; PADAC, Pulmonary-Allergy Drugs Advisory Committee.
Also complicating the analysis was the high differential dropout rate during the trials, leading to problems with missing data analysis. Handling of missing data is difficult, and a National Research Council report summarized in NEJM in 2012 describes the problems associated with various adjustment methods.7,8 In CF-301, 37% of randomized participants withdrew in the DPM arm compared with 28% in control, and in CF-301, withdrawals were less common at 17% versus 12% respectively. Participants who dropped out before week 6 do not contribute any data to the intent-to analysis, and the continued differential dropout rate during the trial resulted in only 66% of DPM and 75% of control patients in CF-301 and 85% and 92% in CF-302, respectively, having data at week 26 for analysis. Various sensitivity analyses were conducted largely confirming the primary analyses, but the high dropout rate raised significant concerns with the safety of DPM.
Data on the episodes of hemoptysis reported as adverse events in the pediatric participants were discussed at length. As stated previously, participants were excluded if they were at risk of hemoptysis defined as >60 mL within 3 months of enrollment and if they failed the mannitol tolerance test at baseline. Thus, the children and adolescents included in the study were presumably at a lower risk for this adverse event. Nonetheless, in the two studies combined, 6.1% (n=4) and 0% of children aged 6–11 years and 9.1% (n=8) versus 3.1% adolescents in the DPM and control groups, respectively, experienced any hemoptysis. This compares with the adult population that had rates of 10.6% and 8.2% in the DPM and control groups respectively. The difference in rates of hemoptysis between DPM and control in pediatric participants was much greater than that seen in adults, which raised concerns about use in pediatric patients. Few of these events were considered severe (one in each pediatric age group receiving DPM and none in control) or were recorded as a serious adverse event (one event in an adolescent on DPM and none in control). While approximately 73% of children in the trials had severe underlying CF and may have been more predisposed to hemoptysis, there was no evidence of an imbalance in the severity between the treatment groups to explain this differential rate in events. In addition, while hemoptysis occurs frequently during exacerbations, the study investigators were not specifically instructed to report each occurrence of hemoptysis as an adverse event. Thus, the occurrences reported as adverse events were likely of a nature that was unexpected in the clinical judgment of the investigator. The PADAC concerns for pediatric use also included the unknown risks of using an airway irritant long term, use in children with milder disease who may have a different risk–benefit profile than the children included in the trials, and the effect of potentially relatively greater adherence, and thus drug exposure, in children when imposed by the parent compared to adults. The two large trials of hypertonic saline provide little additional data on adverse events to help with resolving issues of a safety for inhaled hyperosmolar agents. Indeed, no data were reported on hemoptysis in one trial, and hemoptysis events were captured only in the context of a pulmonary exacerbation in the other, and there were no data reported separately for pediatric patients.9,10
The PADAC was asked to vote on the efficacy and safety when considering use of DPM in the population of children and adults aged 6 years and older. Many members commented on their support for DPM in the adult population and the importance of adding a new class of drug for the management of CF. The vote against approval was largely driven by the equivocal efficacy found in one of the two Phase III trials, the lack of any benefit on the primary endpoint in pediatric participants, and by the safety concerns of hemoptysis in pediatric participants.
Footnotes
Author Disclosure Statement
No competing financial interests exist.