
Abstract
We describe an unusual family in which 2 adult siblings and the son of 1 are all affected by cystic fibrosis with rare mutations and different phenotypes. We discuss the dilemma surrounding the treatment of Pseudomonas aeruginosa colonization in the asymptomatic father, while minimizing disruption to the father–son relationship.
Introduction
C
One factor definitely influencing CF pneumopathy is the presence of respiratory infections caused by microorganisms, such as Pseudomonas aeruginosa, which is a bacterium associated with more rapid lung function decline 5 and reduced survival rates. 6 The age of contracting the infection can vary considerably, and not all patients with CF contract P. aeruginosa, indicating the presence of modifying factors. Several demographic, genetic, and environmental factors have been shown to modify the risk of pulmonary infection with P. aeruginosa. 7
In this article, we describe the heterogeneity of the CF phenotype and its relationship to some rare CFTR mutations in a CF family. We further discuss the role of P. aeruginosa colonization within the same family and the rules to apply to avoid the transmission of pathogen germs among family members of different ages and clinical severities.
Case Report
In July 2012, a 47-year-old male (Patient 1) contacted our facility for assessment following a computed tomography (CT) scan showing bilateral bronchiectasis. This patient's clinical history included chronic lung disease from adolescence with minor hemoptysis since the age of 19 years, infection from Staphylococcus aureus, and azoospermia. Chronic nasal obstruction was also reported and a CT scan showed chronic sinusitis. Gastrointestinal symptoms were absent. Patient 1 was 188 cm tall and his BMI was 24.7 kg/m2. Having sought medically assisted reproduction, he was found at the age of 32 to be a carrier of the G542X CFTR mutation, but no further analysis was advised.
At our facility, sweat testing was conducted twice, according to the Gibson and Cooke method, resulting in chloride levels of 72 and 74 mEq/L. Genetic analysis (reverse dot blot followed by denaturing gradient gel electrophoresis [DGGE] and sequencing) showed the presence of the G542X/711+3A→G genotype. The sputum culture was positive for S. aureus and Burkholderia multivorans. Pulmonary function tests (PFT) showed a mixed restrictive/obstructive picture [forced vital capacity (FVC) 3.79 L (65% of predicted), forced expiratory volume in the 1st second (FEV1) 1.87 L (39%), forced expiratory flow between 25% to 75% of vital capacity (FEF25–75) 0.44 L/s (21%)]. Stool analysis showed the absence of steatorrhea and normal values of fecal elastase. Assessment results justified a diagnosis of CF with pancreatic sufficiency (PS).
Patient 1 also provided us with the clinical history of his younger brother, 42 years old, who was equally azoospermic, but had no symptoms of respiratory disease (Patient 2). He had used in vitro fertilization to bear a son, at which time he was found to be a carrier of the G542X CFTR mutation, but no further analysis was advised.
When scheduling the first admission of Patient 2, his 7-month-old son (Patient 3) presented to the emergency room because of vomiting, loss of appetite, and dehydration. His laboratory tests showed hyponatremia, hypochloremia, hypokalemia, and metabolic alkalosis. After rehydration, a sweat test was performed on different days with resulting chloride levels of 85 and 81 mEq/L. This child was the result of a pregnancy facilitated by assisted reproduction treatment, and the newborn screening for CF was not available in the region where the child was born. Denaturing high pressure liquid chromatography (D-HPLC) analysis and sequencing were performed on the child's mother before the pregnancy to rule out CFTR mutations in her genotype, and no such mutation was found. We later performed DGGE and sequencing of both the mother and the child, and results showed the R334L mutation in the mother and the G542X/R334L genotype in the child (Fig. 1). The child showed no additional symptoms and was well nourished, while stool analysis showed the absence of steatorrhea and normal values of fecal elastase. Cough swab cultures were positive for Haemophilus influenzae. A chest radiograph was unremarkable. Ultimately, Patient 3 received a diagnosis of CF with PS.
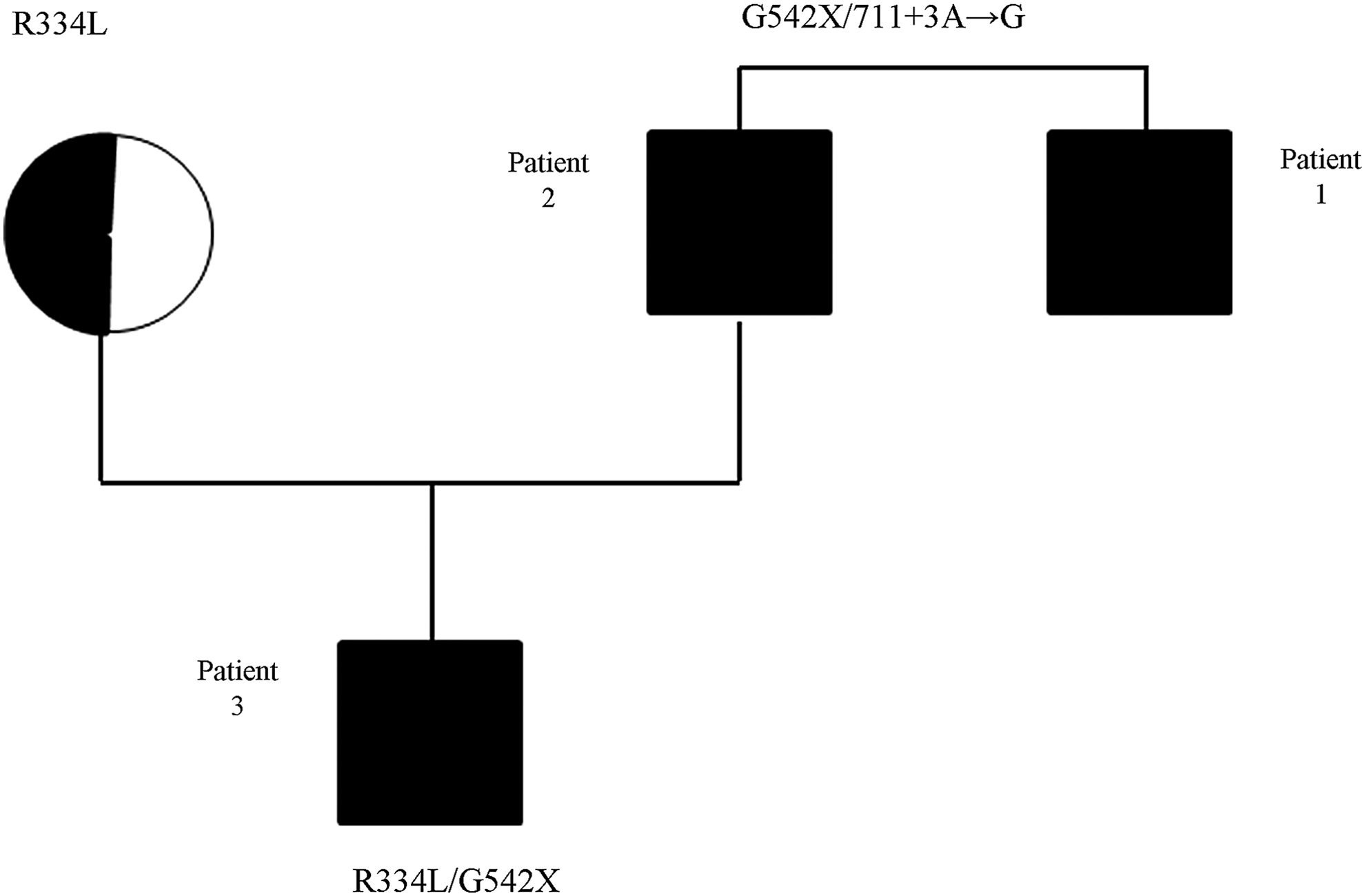
Pedigree of the family.
Thereafter, Patient 2, the father of Patient 3, was evaluated. He reported no symptoms and stood 186 cm tall with a body mass index (BMI) of 29.2 kg/m2. His sweat chloride levels were 78 and 85 mEq/L. The genotype was the same as his brother's (G542X/711+3A→G). Chest radiograph and PFT were normal [FVC 5.47 L (107% of predicted), FEV1 4.37 L (103%), FEF25–75 4.85 L/s (109%)]. Because relevant sinonasal symptoms were not reported, we decided not to perform CT scanning of the sinuses. The patient was unable to expectorate spontaneously, so sputum induction was performed by inhalation of 7% NaCl. The cultures were positive for P. aeruginosa in 2 different samples collected on different days. We then decided to treat with oral ciprofloxacin (1,000 mg bid for 3 weeks) and tobramycin solution for inhalation (TSI) (300 mg bid for 4 weeks). A culture of induced sputum (IS) was repeated 3 weeks after the end of the TSI cycle, and the result was again positive for P. aeruginosa. We decided to perform an additional cycle of antibiotic treatment with oral ciprofloxacin and TIS for 4 weeks. However, the cultures of IS, performed every 2 months for the following 6 months, were persistently positive for P. aeruginosa. We therefore became convinced of chronic colonization, possibly long standing. In the absence of clinical symptoms, we decided not to treat the patient further although we were concerned about the risk of cross-infection to his young son.
We obviously excluded the possibility of intrafamilial segregation, giving utmost preference to the father and son relationship. After discussing the child's risk of P. aeruginosa infection with the parents, we made several recommendations: (1) maintain appropriate hand hygiene; (2) use of a face mask when a cough was present; (3) sleep in different rooms; (4) treat sinks in the home with hypochlorite; and (5) have a strict follow-up of the child with monthly cough swab cultures. At the same time, we strongly advised against close contact between the child and the uncle (Patient 1), as he had evident lung disease and was actively expectorating and carrying S. aureus and B. multivorans in the sputum culture. To date (after 18 months of follow up), the cultures of the child are negative for P. aeruginosa, whereas the cultures of IS of the father remain positive for P. aeruginosa. Both the father and the son still have no respiratory symptoms. The family calmly accepted the hygienic advice and the restrictive rules and adhered to them properly.
Discussion
These case reports document, particularly in the siblings (Patients 1 and 2), the clinical heterogeneity of CF patients with the same genotype. The pancreatic status 8 and fertility 9 in these patients, which strongly correlate with CFTR gene mutations, were obviously concordant, whereas the lung phenotype was strongly discordant. The big difference between the PFT data and the radiographic pictures of the 2 adult patients reinforces the evidence that several factors other than CFTR mutations affect the pulmonary phenotype. 4 Important efforts are underway in North America and Europe to identify further genes of biological relevance to CF. 10
One example is the rare mutation 711+3A→G. This discovery was initially reported in patients of Balkanian origin 11 and is described on the CFTR2 website 12 as a CF-causing mutation associated with PS, as in our patients, and has a high prevalence of P. aeruginosa colonization.
R334L is a missense mutation, described thus far in 1 adult male patient, carrying I336K on the other allele, with congenital bilateral absence of the vas deferens (CBAVD), PS, recurrent pulmonary infections, and nasal polyposis. 13 In our database, however, the mutation R334L has been identified in one other patient, now 6 years of age. This child was diagnosed as CF with PS, on the basis of a positive newborn screening, the presence of the CF-causing mutation 3659delC on the other allele, and a positive sweat test. We can therefore confirm the association of the R334L mutation with increased sweat chloride levels in the sweat test and with PS.
When children or young adults are diagnosed with the genetic potential for CF in the absence of symptoms, physicians attempting to guide such patients and families can benefit from an accurate knowledge of phenotype and of the natural history of patients with rare genotypes who are diagnosed late. In fact, recent data 14 indicate that in patients with the nonclassic form of CF, when diagnosis or treatment is delayed, the morbidity of nonclassic CF may be severe. These findings support the conclusion that asymptomatic or oligosymptomatic individuals identified with a genetic predisposition for developing CF, even if mild, should be monitored regularly at the CF centers for disease progression.
The second main issue addressed in this case report is the dilemma over how best to avoid the unusual risk of cross infection between a father and a son both with CF, considering the high probability of intrafamilial transmission of P. aeruginosa.15,16 There is the additional goal of preventing cross infection with the child's uncle, who was carrying B. multivorans in the sputum culture, a germ less virulent than Burkholderia cenocepacia, but equally deserving of attention as P. aeruginosa. 17 Complete avoidance of patient-to-patient contact is the gold standard to prevent the passage of microorganisms from one individual to another. This strategy along with some further principles (hand hygiene, respiratory hygiene, monitoring of healthcare settings, appropriate cleaning and disinfection of home devices) was stated in the United States Cystic Fibrosis Foundation (CFF) guidelines published in 2003. 18 Recent studies have furthered our understanding of the increased morbidity and mortality associated with CF pathogens. The additional evidence of the importance of infection control in CF 19 prompted the CFF to increase the stringency of its recommendations. 20 Our center adopted the policy of infection control as stated by the Italian CF Society, following a systematic review of the literature and of the existing guidelines,21,22 the main principles of which were integrated. The key strategy in our infection prevention measures is to implement contact precautions for all patients by all healthcare workers, regardless of respiratory tract culture results. Moreover, we have instituted a program of continuing patient education, to learn about germs and how they affect people with CF, as well as how to practice good infection control to keep germs from spreading.
The challenging complexity of our case is the need to reconcile the risk of developing a precocious P. aeruginosa infection in the young child, amidst the unpredictable outcomes of lung health and the long-term prognosis, with the desire to minimize disruption to the father–son relationship. While the absence of respiratory symptoms and of sputum production in the father most likely makes it easier for the child's parents to follow the hygienic rules we gave them, we emphasized the necessity to be rigid in their application because of the high risk of transmission of P. aeruginosa due to the cohabitation. These rules, together with the careful follow-up of the child, including monthly cultures of the respiratory tract sample, are an effective and acceptable choice at present to decrease as much as possible the chance of P. aeruginosa cross infection in this young CF child.
Footnotes
Acknowledgment
The authors thank Mrs. Janette Sloan for her language assistance and editing.
Author Disclosure Statement
No competing financial interests exist for any of the authors.