
Abstract

Introduction
T
Case Report
A 13-year-old male presented with a 6-month history of dry cough and a 1 week history of worsening shortness of breath and pleuritic chest pain. In addition, he had headaches, dry eyes/mouth (Sicca syndrome), and intermittent proximal interphalangeal joint swelling. There was no fever, night sweats, weight loss, skin rash, vision changes, or gastrointestinal symptoms. The family kept a dog and cat as pets, but there was no contact with other animals, construction sites, or cigarette smoke. There was a strong family history of rheumatologic disorders, including scleroderma and dermatomyositis.
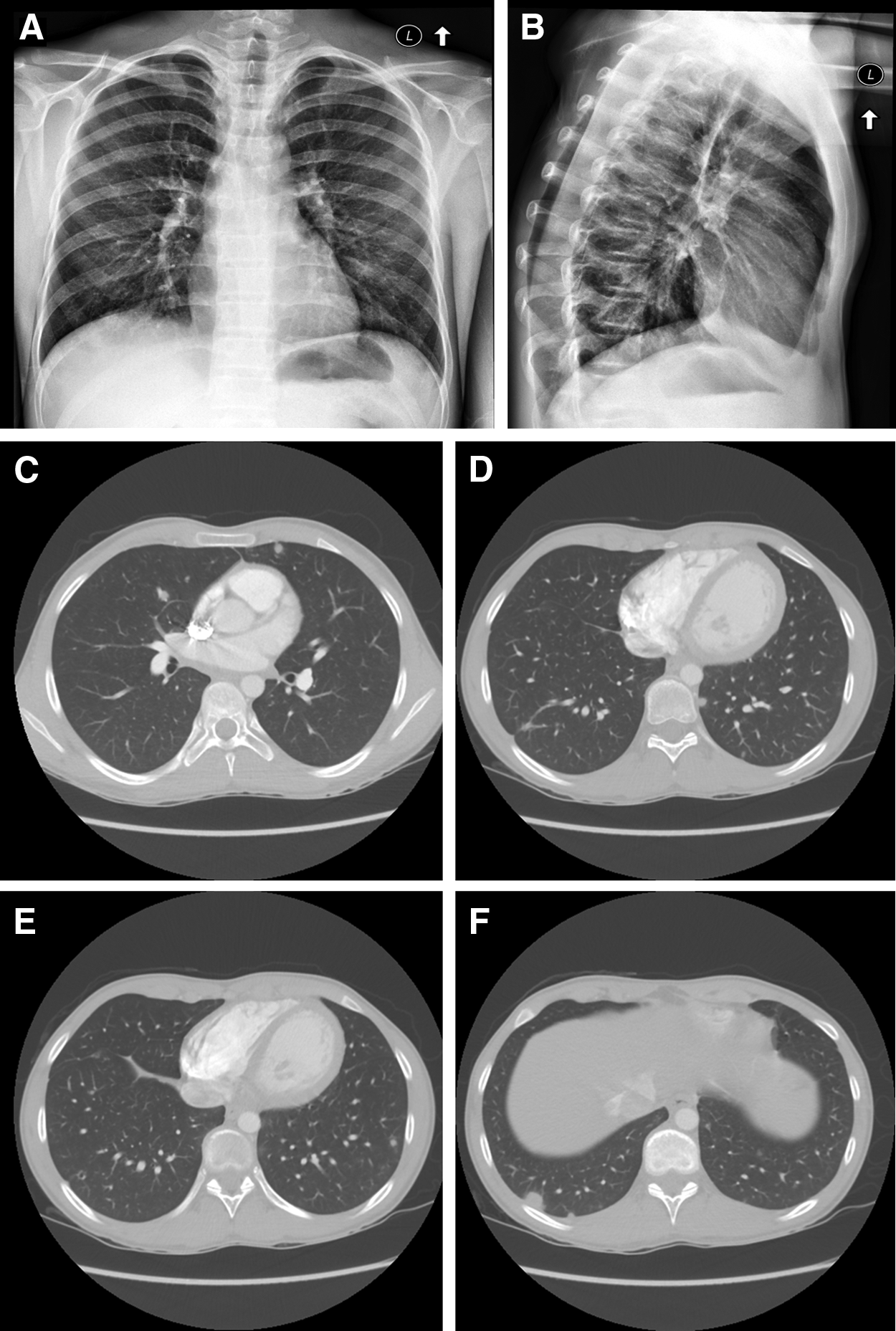
The patient's vital signs were normal, and his weight was at the 63rd percentile. The physical exam was unremarkable except for mild epigastric tenderness. Initial blood tests, including blood count with differential, C-reactive protein, lactate dehydrogenase, uric acid, ferritin, electrolytes, renal and liver function, and urinalysis, were normal. The sedimentation rate was slightly elevated at 18 mm/h (reference range 0–15 mm/h). A chest x-ray demonstrated nodular opacities (Fig. 1A and B); chest computed tomography (CT) confirmed multiple (>40) small, solid, bilateral pulmonary nodules and no lymphadenopathy (Fig. 1C–F). Most nodules were pleural based, and some had central lucency. Serum angiotensin-converting enzyme level was 32 IU/L (reference range 16–65 IU/L). Rheumatologic workup, including antinuclear antibody, anti-double-stranded DNA antibody, antineutrophil cytoplasmic antibody (ANCA), antimyeloperoxidase, and von Willebrand factor antigen, was negative. Fungal serology panel, aspergillus antigen, urine histoplasma antigen, tuberculin purified protein derivative skin test, quantiferon TB, and mycoplasma and chlamydia serologies were negative. He had normal spirometry and plethysmographic lung volumes but a diffusion capacity of 70% predicted. A thoracoscopic wedge resection of two nodules demonstrated exuberant granulomatous inflammation involving the lung and pleura. A mixture of necrotizing and non-necrotizing epithelioid granulomas was present (Fig. 2). Granulomas were noted surrounding airways and vessels and focally invaded small vessel walls, breaking into the lumen (Fig. 3). Special stains for organisms performed on multiple sections, including Ziehl–Neelson, Fite, Grocott's methenamine silver, and Warthin–Starry, were all negative. No foreign material was identified. Bacterial and fungal cultures were negative.
(

Multiple small epithelioid non-necrotizing granulomas composed of lymphocytes, epithelioid histiocytes, and scattered multinucleated giant cells are seen (stars) in addition to one granuloma with central coagulative necrosis (arrow). Color images available online at www.liebertpub.com/ped
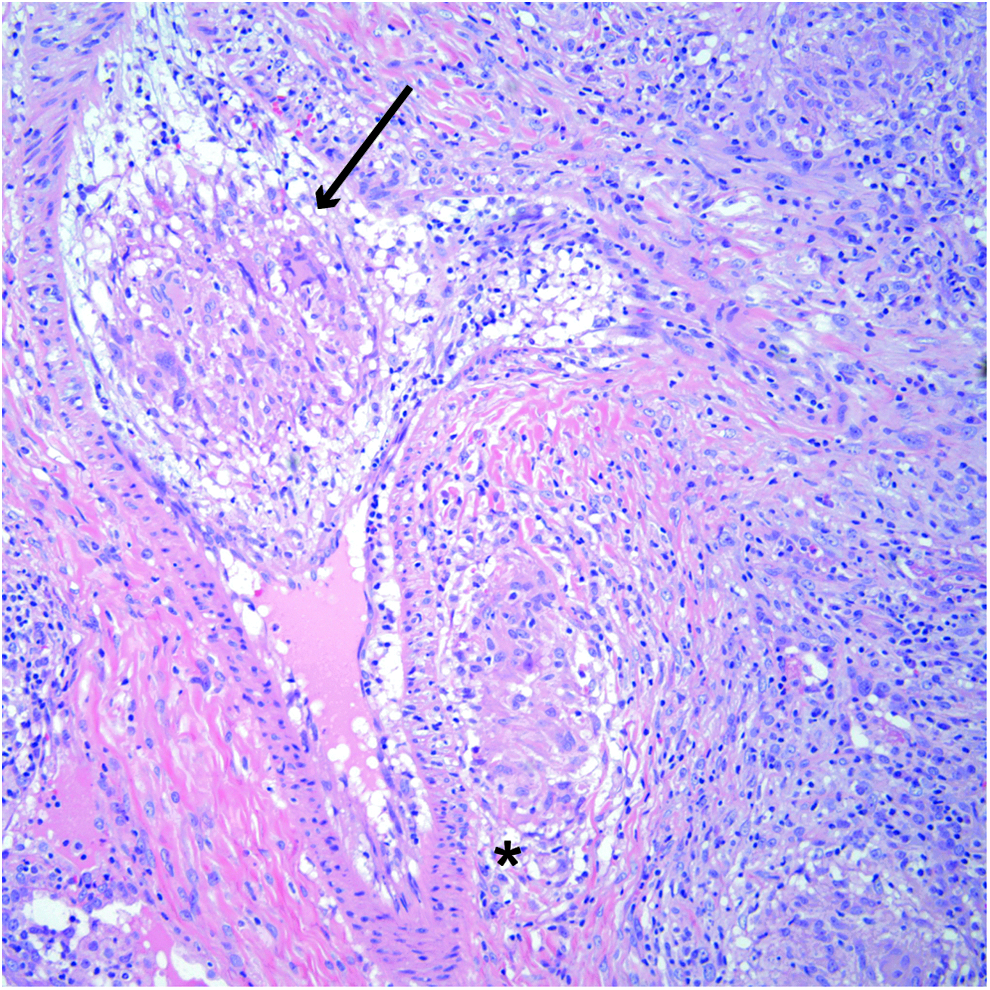
One epithelioid granuloma is present adjacent to a small vessel (star), and one granuloma invaded the vascular wall and broke into the lumen (arrow). Color images available online at www.liebertpub.com/ped
With negative microbiological serology studies, tissue stains, and cultures, infectious causes were ruled out. The presence of vasculitis and extrapulmonary manifestations raised the possibility of a small vessel vasculitis, but the absence of uncontrolled asthma, chronic rhinosinusitis, peripheral eosinophilia, renal disease, or positive ANCA ruled out GPA and EGPA. The clinical, radiologic, laboratory, and histologic findings in the patient were not typical for classic sarcoidosis. The presence of granulomatous vasculitis and necrosis and exclusion of other entities in the differential diagnosis were consistent with necrotizing sarcoid granulomatosis (NSG). NSG is a rare condition characterized by sarcoid-like granulomas, variable degrees of necrosis, vasculitis, frequent extrapulmonary manifestations, and a generally favorable prognosis. 1
The patient continued to have headaches, joint pain, mouth/eye dryness, and chest pain. A head computed tomography (CT)/magnetic resonance imaging (MRI)/magnetic resonance angiogram (MRA) confirmed no cerebral or vascular abnormalities. Given previous case series identifying favorable outcomes in patients with NSG treated with steroids,2,3 the patient received pulse therapy of 1,000 mg intravenous methylprednisolone daily for 3 days, and was subsequently transitioned to oral corticosteroid therapy with prednisone, 60 mg daily. Within 1 month of starting steroids, he experienced symptomatic relief of headaches, dry mouth, and joint pain, as well as normalization of his diffusion capacity. During the sixth week of his steroid wean, a repeat chest CT demonstrated near complete resolution of the nodules, and the sedimentation rate had normalized. Chest pain resolved, although the patient complained of abdominal pain, as well as tenderness in the proximal interphalangeal joints and sacroiliac joint. A pelvic MRI identified irregularity and sclerosis of the right sacroiliac joint consistent with history of sacroiliitis but no active synovial inflammation. He was treated with azathioprine (50 mg daily) as a steroid-sparing agent, and experienced resolution of the arthritis. However, his course was complicated by persistent generalized myalgias and frequent headaches consistent with amplified musculoskeletal pain syndrome and nonsteroidal anti-inflammatory drug (NSAID) overuse.
Discussion
Necrotizing sarcoid granulomatosis was described for the first time in 1973 by Liebow who identified 11 patients with sarcoid-like granulomas, variable degrees of necrosis, and vasculitis. 1 Since then, nearly 100 adult cases and 10 pediatric cases have been reported in the literature.2–11 Extrapulmonary involvement is estimated to occur in nearly 86% of adult patients, and has been reported in half of the pediatric cases reported.2,3 The majority of patients experience resolution of their disease, and some require steroids or immunosuppression, particularly those with more severe systemic or neurologic manifestations.
Necrotizing granulomas have a broad differential, which includes mycobacteria, fungal infections, GPA, and foreign body-type inflammation. Non-necrotizing granulomas are more often a manifestation of a noninfectious cause such as sarcoidosis and vasculitides. The pathology of NSG is unique in that it demonstrates features of both sarcoidosis and granulomatous vasculitides, raising the question of whether this disease represents a variation of sarcoidosis with necrosis of the granuloma and vessels, or a necrotizing angiitis with sarcoid reaction. The cell types involved, microbiological stains, and associated patterns of vasculitis and necrosis provide useful clues into the underlying cause.
NSG shares several pulmonary and extrapulmonary clinical features with sarcoidosis, including a generally favorable prognosis2,3,12 and similar immunopathologic 7 and histopathologic findings.13–15 In fact, an estimated 5% of granulomas in sarcoidosis show the typical histology of NSG, including necrotizing granulomas.15,16 Lazzarini et al. reported a 15-year-old girl with NSG and a strong family history of sarcoidosis, and suggested a possible autoimmune link between the two diseases. 8 However, the peripheral location of the nodules, lack of hilar adenopathy, and normal angiotensin converting enzyme would suggest that NSG and sarcoidosis are distinct entities. NSG shares some pathologic features with granulomatous angiitides, but is not associated with an elevated ANCA, renal disease, or asthma.
The differential diagnosis of pulmonary granulomatous diseases in children is broad and requires a thorough evaluation for hematologic, infectious, immune-mediated, and vasculitic disorders. To our knowledge, this patient is the eleventh pediatric case of NSG reported in the literature. NSG is an example of a disease with unclear classification, as some of its features overlap with other granulomatous and vasculitic disorders.
Footnotes
Author Disclosure Statement
No competing financial interests exist.