
Abstract
We performed cytokine profiling in a familial case with co-mutations of FAS and MEFV. The child was referred to our hospital because of splenomegaly. Her mother was hospitalized at a young age because of frequent fever and lymphadenopathy. However, despite several examinations, including bone marrow evaluation, her mother's condition was undiagnosed. Her frequency of fever decreased with age. The number of double-negative T cells increased in both mother and child cases. Their number of apoptotic cells cultured under phytohemagglutinin and interleukin (IL)-2 did not change with the anti-Fas antibody concentration. The genomic study of the proband and mother revealed co-mutations of FAS as p.Leu229fs on exon 9, which is located in the death domain in the intracellular signal pathway, and compound heteromutations as p.L110P and p.E148Q on exon 2 of MEFV. Soluble IL-2R was constantly high in both mother and child cases. Serum IL-10, MCP-1, and MIP-1β levels were high only in the child with recurrent fever. The mother showed a low level of these cytokines, but a relatively high level of IFN-γ. From their history and data, the immunological status, which is correlated with familial Mediterranean fever, might influence apoptosis in autoimmune lymphoproliferative syndrome.
Introduction
A
In this report, we present the case of a mother and her child with ALPS diagnosed on the basis of splenomegaly, whose mutations were found in FAS and MEFV which cause familial Mediterranean fever (FMF). Their cytokine profiling results and numbers of double-negative T cells were variable. Their symptoms gradually improved with age.
Case
The patient was born as an appropriate for date preterm (3,344 g at 39 weeks). At 1 year of age, she was referred to our department because of splenomegaly diagnosed when she was brought to a clinic for diarrhea, rash, and fever in August 2014. Her past history was unremarkable.
Her mother and mother's brother also had histories of splenomegaly. The mother's brother was not analyzed genetically. Moreover, her mother had a history of frequent fever and urticarial-like erythema at a young age. Bone marrow examination could not establish a definitive diagnosis. These episodes were reduced and have become almost nonexistent recently.
The child's height was 83 cm (−0.1 SD) and her weight was 11.2 kg (+0.4 SD). Her physical examination results were as follows: body temperature, 36.6°C; heart rate, 128/min; blood pressure, 102/58 mmHg; respiratory rate, 32/min. She had no facial abnormality or lymph node enlargement. Her liver was not palpable. Her spleen was palpable at 2.5 cm below the costal margin. Petechia was found in her lower extremities. Ultrasonographic findings revealed splenomegaly (10 × 6 cm).
The child's blood cell profile was as follows: white blood cell count, 15,800/μL (neutrophils 31%, lymphocytes 62%, atypical lymphocytes 2%, and monocytes 2%); hemoglobin level, 10.5 g/dL; platelet count 20.2 × 103/μL. Her liver and kidney function tests were both normal. Her C-reactive protein level was 0.22 mg/dL. Her IgG, IgA, IgM, and IgD levels were 2,730, 95, 124, and 1.6 mg/dL, respectively. Anti-cytomegalovirus and Anti-Epstein-Barr virus antibodies were all negative. Her serum soluble interleukin (IL)-2R level was extremely high at 10983.0 U/mL as shown in Table 1. Her IL-10 level was also high at 170.48 pg/mL.
Bold values showed higher than normal levels.
ALT, alanine transaminase; ANA. anti-nuclear antibody; APTT, activated partial thromboplastin time; AST, asparate transaminase; BUN, blood urea nitrogen; CK, creatine kinase; CMV, cytomegalovirus; CRP, C-reactive protein; EBV, Epstein-Barr virus; LDH, lactate dehydrogenase; PT-INR, prothrombin time-international normalized ratio; TP, total protein; WBC, white blood cell.
Three-color flow cytometry revealed increased proportion of double-negative T cells (TCRαβ+CD4−CD8−) at 13.75%, as shown in Table 1.
Peripheral blood mononuclear cells were stimulated with phytohemagglutinin at 10 μg/mL, IL-2 at 100 U/mL for 4 days, and IL-2 at 50 U/mL for 10 days. The annexin V+, PI−, and PI+ T cell percentages and DNA content analysis of cultured cells showed no significant changes after treatment with the different concentrations of anti-Fas antibody for 12 h at 37°C compared with the control (Figs. 1 and 2).7,8
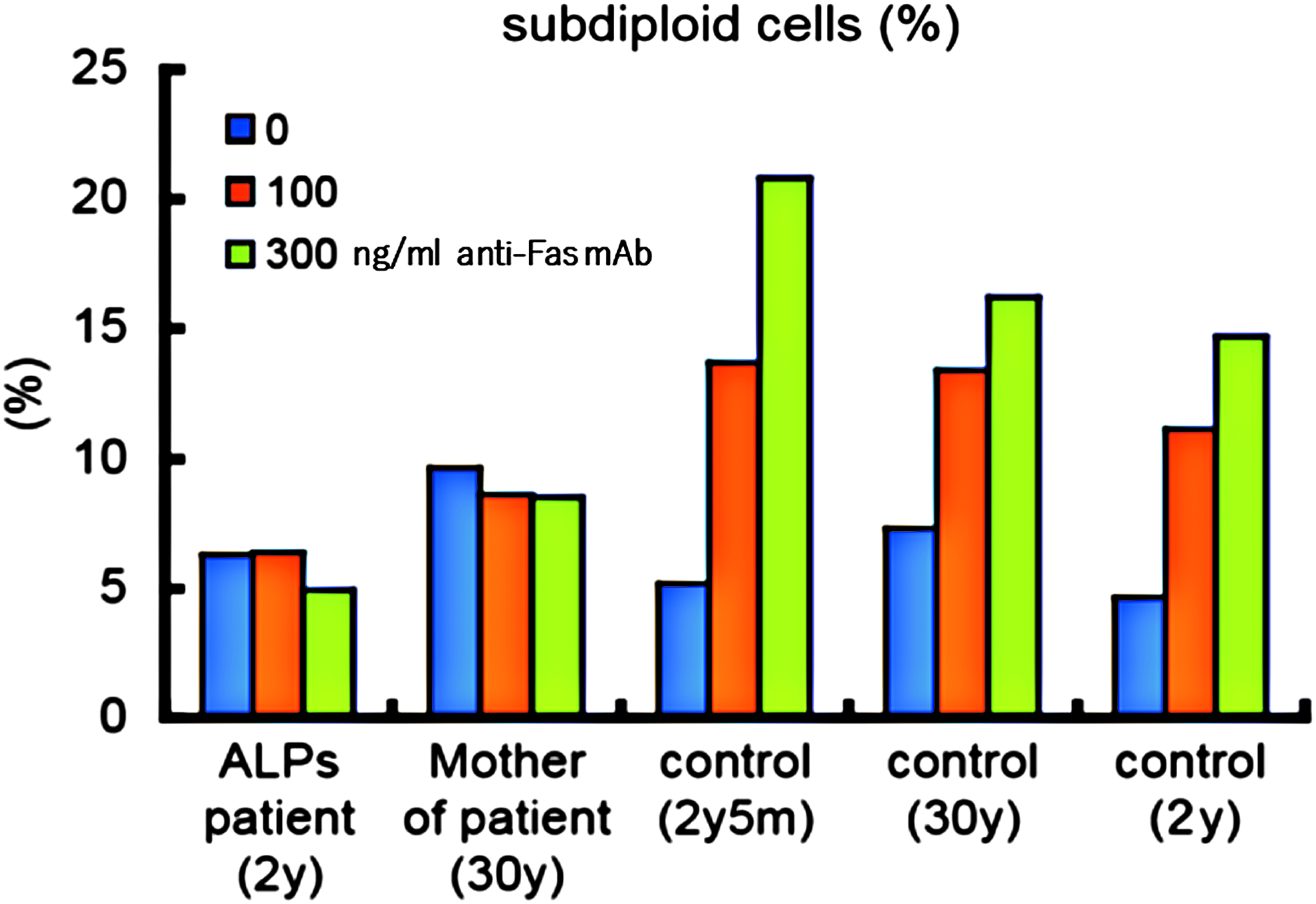
Apoptosis (DNA content analysis) induced by anti-Fas antibody at an incubation time of 12 h at 37°C in lymphocytes cultured under 10 μg/mL PHA and 100 U/mL IL-2 for 4 days followed by 50 U/mL IL-2 for 10 days. IL, interleukin; PHA, phytohemagglutinin.

Apoptosis induced by anti-Fas antibody at an incubation time of 12 h in Annexin V+ PI+ T cells and Annexin V+ PI− T cells, which were cultured under 10 μg/mL PHA and 100 U/mL IL-2 for 4 days followed by 50 U/mL IL-2 for 10 days
Her IL-1, IL-2, IL-4, IL-6, IL-12, IL-17, G-CSF, GM-CSF, IFN-γ, and TNF-α levels were less than the detectable levels. Her IL-8 and IL-13 levels were within the normal ranges at 4.1 and 0.53, respectively. The MCP-1 and MIP-1β levels were relatively high at 25.43 and 184.49 pg/mL, respectively (Fig. 3).
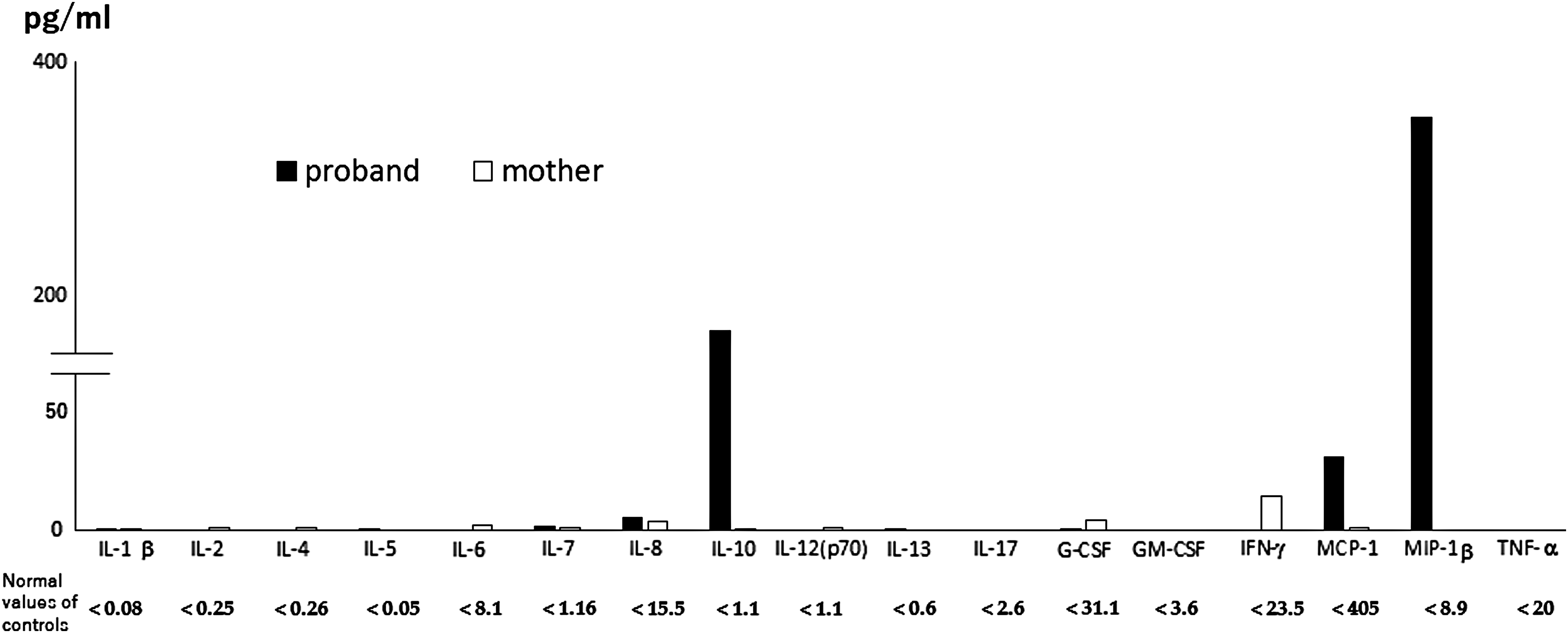
Serum cytokine profiling using Multiplex Cytokine Bead assay (BioRad, USA). Control values were determined using normal healthy volunteer controls.
The mother's serum soluble IL-2R level was also high at 1061.0 U/mL (Table 1). The IL-10 and IL-17 levels were not increased. The proportion of TCRαβ+CD4−CD8− cells was high at 8.02%. Apoptosis induced by anti-Fas antibody was suppressed similarly as her daughter. Her serum cytokine profiling showed quite a different pattern, that is, high IFN-γ level without increased levels of other cytokines (Fig. 3 and Table 1).
In the genomic study of the child and her mother, FAS showed a frame shift as p.Leu229fs (c.685_686del) on exon 9, which is located in the death domain in the intracellular signal pathway. There were no mutations of KRAS, NRAS, FASL, SH2DA1, CASP8, CASP10, TNFRSF6B, and SPP1 by Ion Proton whole-genome exome sequencing. MEFV showed compound heteromutations as p.L110P (T329C) and p.E148Q (G442C), which corresponded to the causative mutations of MEFV already reported. 9 The mother also showed p.L110P (T329C) heteromutation and p.E148Q (G442C) homomutation.
The child showed periodic fever and symptoms of infections as confirmed by leukocytosis in blood examinations every 2 to 3 months. The frequency of these episodes decreased until 3 years of age. However, her soluble IL-2R continued to be high. Her splenomegaly gradually subsided. The mother and child now receive regular blood checks. The levels of soluble IL-2R of the child are still high at more than 6,000 U/mL.
With regard to histories of splenomegaly of the mother and her brother, the family genetic pedigree is shown in Fig. 4.
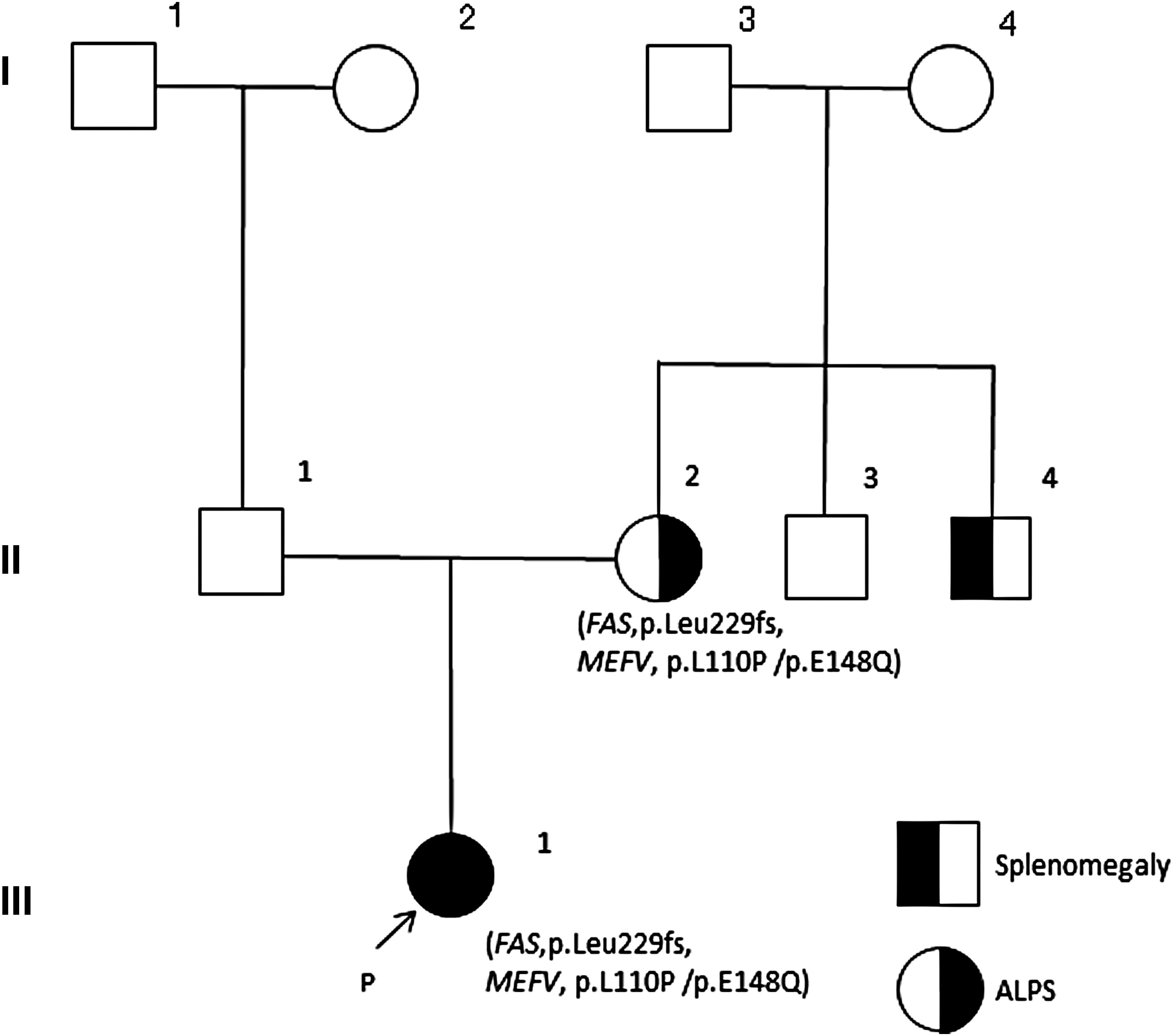
Family genetic pedigree.
Discussion
ALPS, a disease first described by Sneller et al., is characterized by chronic lymphadenopathy, hepatosplenomegaly with alterations in gamma globulins, and manifestations of autoimmune diseases. 10 The initial onset of these cases ranges from 3 months to 3 years of age. Shortly thereafter, the pathogenesis of large numbers of an unusual CD4−CD8− T cell population in peripheral blood and lymph nodes, and dysfunction of apoptosis were clarified in these cases. The unusual CD4−CD8− T cell population is called “double-negative T cells,” and several genomic or somatic genomic mutations of the FAS-related signal pathway have been known in most cases. Type Ia ALPS, which is caused by FAS mutation, is the most common type. In this report, the patients have hetero missense mutation in the death domain of FAS. The mutation coincided with an already reported case [FAS 05185(DB-ID)] in an NIH ALPS program (www.ncbi.nlm.nih.gov/lovd/home.php?select db = FAS). Both familial cases have shown increased number of double-negative T cells and dysfunction of apoptosis as induced by anti-FAS antibody. From these data, both cases convincingly meet the criteria of ALPS. Interestingly, the cases had another mutation of MEFV, which was suspected to be the cause of FMF. These mutations are affected by a dominant-negative effect in the signal pathway which is correlated with pyrin.
The mutation of the nondeath domain was suspected by another mechanism called haploinsufficiency. 11 Coinherited mutations of FAS and caspase-10 were reported by Cerutti et al. 12 Also, polymorphisms in the FAS, FASL, CASP8, and SH2D1A genes were reported to be correlated with susceptibility to ALPS. 13 Another report in the literature showed that mutations causing ALPS have been influenced by mutations involved in lymphoproliferative diseases and familial hemophagocytic lymphohistiocytosis (PRF1 and UNC13D genes).14,15 However, we analyzed these mutations using whole exosome sequence and did not find any pathogenic mutation in these genes, except MEFV.
MEFV coding for pyrin regulates the production of IL-1β and activates NF-κB through inhibition of the reaction between cryopyrin and ASC, and suppresses inflammation. 16 Inflammasome consists of 3 complexes of caspase 1, NLRP, and ASC. Pyrin suppresses the activation of inflammasome by competing for ASC. Serum IL-1β, which represents the activation of inflammasome, was detected only in the mother. Therefore, increased serum IL-1β might have an effect on ALPS symptoms. On the other hand, Christiansen et al. have recently reported a Caucasian 16-year-old boy who had co-mutations of both XIAP and MEFV. The patient complained of recurrent febrile episodes with extensive splenomegaly, lymphadenopathy, and anemia similarly to ALPS. XIAP expression was markedly reduced, whereas a functional assay assessing the TNF-α production of monocytes in response to NOD2 stimulation showed reduced activity. Christiansen et al. suggested that the heterozygous MEFV variant and the hemizygous XIAP variant combination triggered the prolonged and pathological inflammatory response. 17
The clinical signs of the familial case are very mild and decreased with age. The mother showed no increased IL-10 level and did not have any other symptoms with advancing age. ALPS patients who developed lymphoma showed FAS gene mutations. 18 The mother and proband showed high levels of soluble IL-2R, which usually indicates the presence of lymphoma or the activation of T cells. In FMF, Rimar et al. have suggested an increase in the number of regulatory T cells after FMF attacks reaching a maximal level at 7 days, and Treg cells may have a role in terminating FMF attacks. 19 IL-10, which is increased in ALPS, is known to activate Treg cells. Therefore, Il-10 might reduce or suppress the FMF attack. However, the level of cytokines, including IL-10 in the mother with the same mutations, was not high except for IFN-γ.
FMF is accompanied by an increased serum IL-6 level, and is controllable by IL-1-targeting drugs. In combination, IL-6 and IL-1 are known to be potent inducers of Th17 cell development. Ovadia et al. reported that the Th17 population in peripheral blood mononuclear cells in FMF patients was higher than that in peripheral blood mononuclear cells in healthy subjects. 20 It is known that IL-17 protects T cells from apoptosis and contributes to the development of ALPS-like phenotypes. Patients with ALPS have high serum levels of IL-17 upon activation in vitro. IL-17 neutralization substantially increases Fas-induced cell death in T cells from ALPS patients in vitro. Treatment with anti-IL-17 antibodies ameliorates the autoimmune manifestations and prolongs survival in an animal model of ALPS. 21
Further investigations of penetrance without symptoms, including cytokines, the T cell population, and genomic studies, are needed to make a definitive conclusion.
Footnotes
Acknowledgments
The authors thank Dr. Edward Barroga (![]() ), Associate Professor and Senior Medical Editor from the Department of International Medical Communications of Tokyo Medical University for editing the article. This work was not supported by any grants, and the authors have no conflicts of interest associated with this study. They received written permission from the parents to publish their deidentified information and that of their child in this report.
), Associate Professor and Senior Medical Editor from the Department of International Medical Communications of Tokyo Medical University for editing the article. This work was not supported by any grants, and the authors have no conflicts of interest associated with this study. They received written permission from the parents to publish their deidentified information and that of their child in this report.
Author Disclosure Statement
No competing financial interests exist.