
Abstract
The treatment of severe combined immunodeficiency (SCID) is immune reconstitution using hematopoietic stem cell (HSC) transplantation early in life. HLA-identical related donors are the preferred source of HSCs. Since sibling donors are available in <30% of patients, other sources of HSCs are considered—mismatched related donor, umbilical cord blood (UCB), and matched unrelated donor bone marrow. We report the outcome of 10 patients with SCID or combined immunodeficiency (CID) 10 years after UCB transplantation (UCBT) at our institution using a retrospective chart review. Eight patients were alive 10 years post-transplantation. This was the second transplant for 2 patients due to initial transplant engraftment failure. Immunologic reconstitution was demonstrated after transplant with presence of memory T cells at 3 months, naive T cells at 12 months, B cells at 3 months, and normal tetanus/diphtheria toxoid antibody responses at 2 years. Immune response remained robust 10 years post-transplantation. Eight patients developed stage I acute graft-versus-host disease (GvHD), 2 patients developed grades 2—4 GvHD, and 1 child developed chronic GvHD with bronchiolitis obliterans. UCB should be considered as an alternative HSC source for patients with SCID and CID because of its robust and sustained recovery of immune function, low risk of severe GvHD, and accessibility.
Introduction
S
Our group previously reported immune reconstitution using UCB transplantation (UCBT) in patients with SCID and CID 6 ; here we report the results of a 10-year follow-up on 10 patients who received unrelated UCBT for SCID or CID.
Materials and Methods
Patients
A retrospective chart review, approved by the Institutional Review Board, was conducted on patients with SCID or CID who presented to Cardinal Glennon Children's Hospital at Saint Louis University between January 1996 and July 2005 and received unrelated UCBT due to the unavailability of a HLA-matched donor for HSCT. Long-term clinical outcomes were assessed for patients >10 years post-transplantation.
Preparative regimen and graft-versus-host disease prophylaxis
Patients CG01 to CG07 underwent myeloablative conditioning (MAC) with busulfan, cyclophosphamide and antithymocyte globulin (ATG). Patient CG03 with reticular dysgenesis (RD) underwent initial UCBT with preparative regimen of MAC; however, the patient failed engraftment and had early trilineage autologous recovery of hematopoiesis. This patient underwent salvage preparative conditioning with fractionated total body irradiation, cyclophosphamide, and ATG followed by a second UCBT from a different unrelated donor. Patient CG08 with autosomal recessive (AR) T–B–SCID initially underwent an allogeneic sibling donor HSCT with preconditioning of fludarabine and ATG, but failed to engraft. This patient underwent a second transplant with unrelated UCBT and MAC consisting of busulfan, cyclophosphamide, and ATG. Patients CG09 and CG10 underwent reduced intensity conditioning (RIC) with fludarabine, ATG, and cyclophosphamide.
Prophylaxis for acute graft-versus-host disease (GvHD) included continuous cyclosporine A and methylprednisolone. CG01 also received methotrexate for GvHD prophylaxis. Patients were evaluated daily during hospitalization for signs of acute GvHD and at least weekly after discharge for the first 100 days post-transplantation. Acute GvHD was diagnosed by clinical evaluation. Corticosteroids were discontinued by day +60 post-transplantation and cyclosporine A was discontinued between day +100 to +365, depending on clinical evidence of GvHD. The patients also received trimethoprim/sulfamethoxazole and fluconazole prophylaxis.
Engraftment
Hematologic recovery was closely monitored and assessed. Neutrophil recovery was defined as absolute neutrophil count (ANC) ≥500 cells/mm3 on 3 consecutive days. Recovery of platelets was achieved when platelets were ≥20,000 cells/mm3 for 7 consecutive days without transfusions. T cell engraftment occurred when CD3+ cells were ≥1,000 cells/mm3. Donor chimerism was determined by restricted fragment length polymorphism (RFLP) analysis or XX/XY fluorescent in situ hybridization (FISH) for sex-mismatched donor–recipient pairs once peripheral blood neutrophil and lymphocyte numbers recovered, usually between 1 and 6 months post-transplantation.
Results
Patient characteristics
Ten patients were identified who received unrelated UCBT for primary T cell immunodeficiency disorders at our institution from January 1996 through July 2005 (Table 1). Patients CG01 through CG05 and CG07 were previously reported after their second year post-transplantation. 6 Three patients had an AR form of SCID (AR SCID), 1 patient had Janus kinase 3 deficiency SCID, 1 patient each had common gamma chain (γC) deficiency SCID, interleukin-7 alpha chain receptor (IL-7Rα) deficiency. Of the patients with CID, 1 patient had RD and 1 patient each had Nezelof's syndrome and Rothmund Thomson syndrome.
γc, common gamma chain; A/W, alive and well; AR, autosomal recessive; CID, combined immunodeficiency; CMV, cytomegalovirus; F, female; FTT, failure to thrive; IL-7Rα, interleukin-7 receptor subunit alpha; Jak3, Janus Kinase 3; LLL, left lower lung; M, male; NB, newborn; PJP, Pneumocystis jirovecii pneumonia; RSV, respiratory syncytial virus; SCID, severe combined immunodeficiency; UCBT, umbilical cord blood transplantation.
All patients had pretransplant immunologic evaluations (Table 2), which demonstrated decreased absolute lymphocyte count (ALC) and decreased/absent T cells except patient CG07 who had CID. CD56+ natural killer (NK) cell numbers were normal in the patients with Nezelof's syndrome (CG01), RD (CG03), CID (CG07), and 2 of the 3 patients with AR SCID (CG04, CG05); however, only 1 of the AR SCID patients had elevated NK cytolytic function (CG05). All patients had decreased T cell lymphocyte proliferation assay (LPA) responses to antigens (Candida albicans, tetanus toxoid), alloantigen stimulations, and mitogens [PHA, concanavalin A (Con A), pokeweed mitogen (PWM)]. CG01 had normal immunoglobulin levels at 13 months old. CG02, CG05, and CG09 had absent or decreased immunoglobulins. CG07 had low–normal IgG 307 mg/dL (normal age range 268–717 mg/dL) with normal IgA, IgM, and IgE. CG10 (IL-7Rα deficiency SCID) had decreased IgG 93 mg/dL (normal age range 196–525 mg/dL), absent IgA, and low–normal IgM level 41 mg/dL (normal age range 39–92 mg/dL).
Con A, concanavalin A; HiB, Haemophilus influenzae type B; MLC, mixed lymphocyte culture to B cell alloantigens; NK, natural killer; NR, normal range; PHA, phytohemagglutinin; PWM, pokeweed mitogen; SD, standard deviation; SI, stimulation index; UCB, umbilical cord blood.
Hematopoietic engraftment
All patients achieved neutrophil recovery post-transplantation, including the 2 patients who died post-transplant. The median time for neutrophil recovery was 10.5 days (Table 3). The 8 patients who survived transplantation achieved stable engraftment of platelets at 34.5 days.
World Health Organization growth charts used to calculate weight and height percentiles for 0–24 months of age. Centers for Disease Control growth charts used to calculate weight and height percentiles for 2 years and older.
Patient CG03 weight and height for post-transplantation documented at 24 months.
%ile, percentile; ANC, absolute neutrophil count; CD, cluster of differentiation; DRmol, molecular HLA-DR; GI, gastrointestinal; HLA, human leukocyte antigen.
Graft-versus-host disease
All but two patients developed acute GvHD grade 1 (Table 3). CG09 (CID and Rothmund Thomson syndrome) developed grade 4 involving skin and gut, which presented as generalized erythroderma, bullae, desquamation, and severe abdominal pain without an ileus. CG08 developed acute GvHD grade 1 of the skin and grade 2 involving the gastrointestinal tract, and subsequently died 32 days post-transplant from complications of gut GvHD and adenoviral sepsis. CG07 with CID and interstitial lung disease (ILD) from multiple lung infections pretransplant developed chronic GvHD with pulmonary fibrosis 6 months post-transplantation. This progressed to bronchiolitis obliterans diagnosed 13 months post-transplant.
Lymphoid development
Overall, ALC increased within the first 3 months post-transplantation, primarily due to expanding T cell population. ALC continued to increase until 60 months where it remained within an age-adjusted normal range (AANR) at 120 months post-transplantation (2,603 ± 601 cells/mm3). CD3+ T cells increased within 3 months post-transplantation and stabilized within an AANR through 120 months post-transplantation (1,797 ± 455 cells/mm3) (Fig. 1). CD4+ T cells did not significantly increase until 12 months post-transplantation but achieved a level within the AANR (1,042 ± 266 cells/mm3) through 120 months post-transplantation (Fig. 1). Within 3 months post-transplantation, CD8+ T cells increased and maintained within the AANR at 120 months post-transplant (718 ± 166 cells/mm3) (Fig. 1). LPA for mitogens PHA and Con A was ≥50% normal response and alloantigen stimulation was ≥30% normal response 3 months post-transplant, which remained stable (Fig. 2A). LPA for PWM reached normal response of ≥50% at 6 months post-transplant and remained in the normal range. LPA for antigens C. albicans and tetanus toxoids reached and maintained a normal stimulation index of ≥3 by 24 and 12 months post-transplantation, respectively (Fig. 2B). Patients did not receive immunizations until off intravenous immunoglobulin (IVIG), which was usually 12 months post-transplantation.
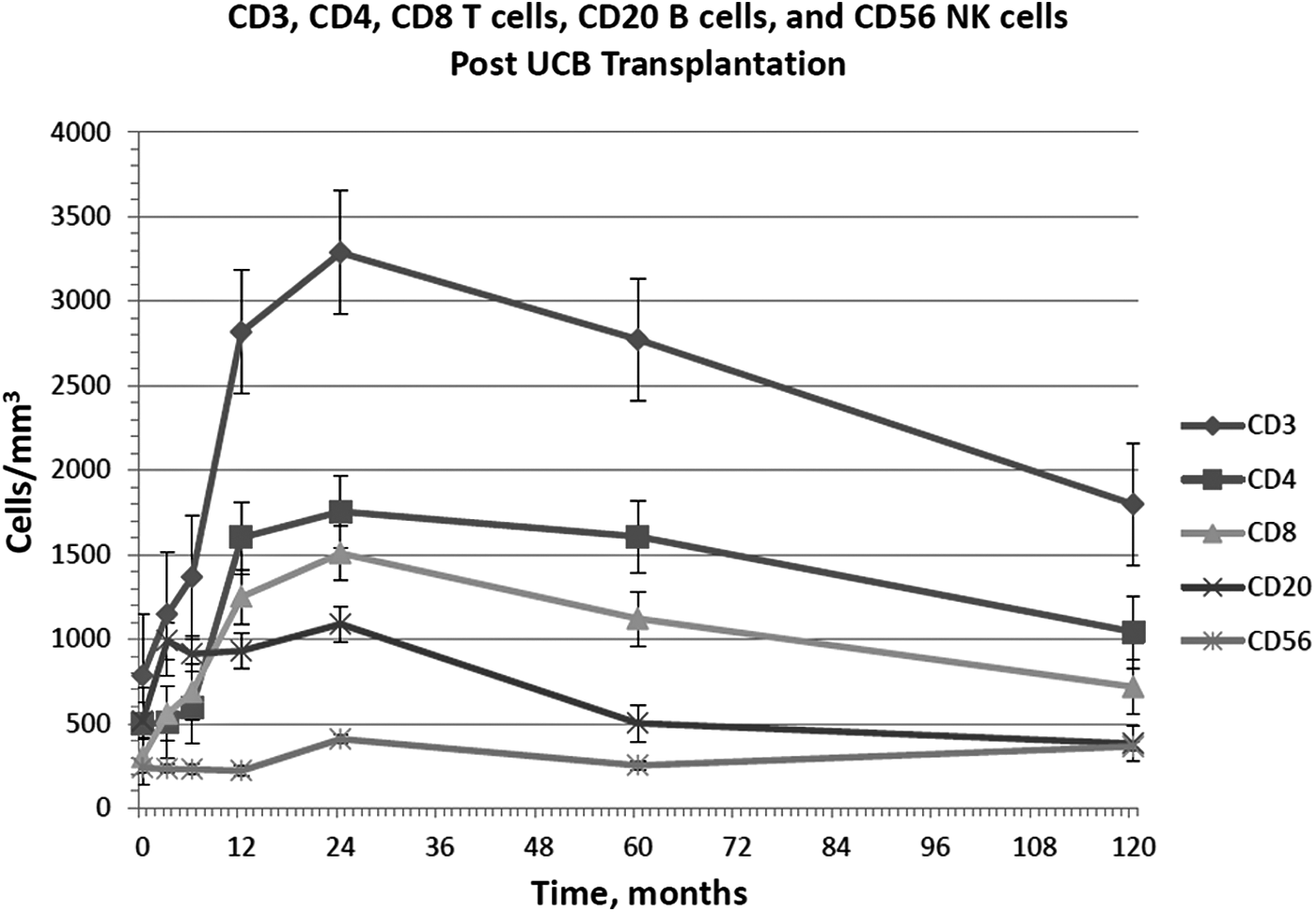
Number of T-, B-, NK cells after UCBT. Data present and mean ± SE. CD, cluster of differentiation; NK, natural killer; UCBT, umbilical cord blood transplantation.

CD3+CD45RA+ and CD4+CD45RA+ markers were evaluated to assess thymopoiesis for the reconstitution of naive T cells. Naive T cells began to increase by 12 months post-transplantation and continued to increase until 60 months with stabilization in the AANR of 64% ± 11.9% (Fig. 3). Memory T cells were determined by measuring CD3+CD45RO+ and CD4+CD45RO+ levels to assess whether expansion of memory T cells occurred post-transplantation. Memory T cells significantly increased in the first 3 months after UCBT (19.8% ± 24.2% versus 65.8% ± 29.3%); however, the percentage decreased by 12 months post-transplantation (19.9% ± 13.3%), then increased again at 60 months (Fig. 3). The memory T cells 10 years post-transplantation were 60.7% ± 21.7%, which is greater than the AANR.

Percentage of naive and memory T cells after UCBT. Data present and mean ± SE.
CD20+ B cells increased within 3 months post-transplantation (Fig. 1). All patients received IVIG for a minimum of 1 year post-transplant. IgG levels remained within the normal range after IVIG was discontinued in all patients except CG07 who was receiving IVIG 10 years post-transplant (Fig. 4). Both IgA and IgM were in the AANR 10 years post-transplant at 142 ± 53 mg/dL and 99 ± 42 mg/dL, respectively (Fig. 4). CG09 was restarted on IVIG 7 years/7 months after UCBT due to hypogammaglobulinemia and recurrent otitis media, which was discontinued 3 years later without development of recurrent infections.

Immunoglobulin levels after UCBT. Data present and mean ± SE. Ig, immunoglobulin.
Antibody responses were documented 24 months post-transplantation in 6 patients to diphtheria and tetanus and in 3 patients to Haemophilus influenzae type B (HiB). All had normal antibody responses to these vaccinations. Only 1 patient had documented Streptococcus pneumoniae antibody titers at 24 months post-transplant with 50% protection (CG10). At 120 months post-transplantation, 4 patients had documented diphtheria and HiB antibody titers. Of these patients, only 3 had normal responses, whereas CG03 had poor antibody titers to both vaccinations. Three patients had documented normal antibody titers to tetanus. Four patients had documented S. pneumoniae antibody titers at 120 months post-transplantation: only 1 of these patients had protective titers (≥1.3 μg/mL) to 75% of the serotypes (CG03). Two of the 4 patients had protective titers to 52.2% of the serotypes (CG09, CG10). At 148 months post-transplantation and 1 month after receiving Pneumovax®, CG09 had protective titers to 82.6% of the serotypes. CG01 had protective titers to 26.1% of the serotypes at 120 months post-transplant.
Five of the 8 patients had documented 100% unsorted donor chimerism by 3 months post-transplantation. CG03 had no engraftment 1 month after the first UCBT but responded with 100% unsorted donor chimerism 17 days after the second UCBT. Two of the 8 patients (CG04, CG09) achieved 100% unsorted donor chimerism 1 year post-transplantation. CG10 achieved only 20% unsorted donor chimerism that remained at 1 and 10 years post-transplantation with normal T cell function.
TRECs were measured in 2 patients. CG01 (Nezelof's syndrome) had 8,312 copies/106 cells 16 years post-transplant. Patient CG10 (IL-7α deficiency) had 6,444 copies/106 cells and 8,974 TREC/106 cells at 9 and 11 years post-transplant, respectively.
Infections
Five of the 10 patients had infections pretransplant (Table 1), including Pseudomonas necrotizing pneumonia requiring left lower lobe lobectomy, respiratory syncytial virus pneumonia, cytomegalovirus (CMV) pneumonia, Pneumocystis jiroveci pneumonia (PJP), Enterobacter cloacae pneumonia, and influenza type A. Some patients had documented infections occurring post-transplantation. CG01 developed Varicella zoster 60 months post-transplantation, which did not require hospitalization. CG02 had sinusitis 60 months post-transplantation. CG03 who required 2 separate transplants had a dental abscess at 120 months after the second transplantation. CG04 had documented positive blood cultures with Staphylococcal aureus at 6 months post-transplantation. Recurrent otitis media occurred in CG09 7 years post-transplantation, requiring myringotomy and tympanostomy tube placement. Pretransplant, CG07 had ILD, CMV pneumonia, and PJP. At 6 months post-transplantation, this patient developed pulmonary fibrosis, which progressed to bronchiolitis obliterans and pulmonary hypertension (chronic GvHD) at 13 months post-transplantation. By 120 months, this patient had other infections including parainfluenza, bronchitis, nonanthracis Bacillus line infection, and PJP.
Growth
Pretransplant, 50% of the patients were below the third percentile for weight with a mean percentile of 19.36. At 120 months post-transplantation, all patients' weights were more than the third percentile with a mean percentile of 45.7 (Table 3). With regard to stature, 6 of the 10 patients' height pretransplant were less than the third percentile (Table 3). Only 3 of the 8 patients 120 months post-transplant were less than the third percentile for height. An exception to the data is CG03 who did not have a documented weight and height 10 years post-transplantation. The data from 24 months post-transplantation were included as 6.1 percentile for weight and 0.6 percentile for height.
Overall survival and cause of death
Eight of the 10 patients were alive 10 years after UCBT. CG06 (γC deficiency SCID) died from cardiac arrhythmia and presumed cardiomyopathy 19 days post-transplantation. The autopsy demonstrated mononuclear and lymphocytic infiltration in necrotic cardiac areas. CG08 (AR T–B–SCID) died 32 days after the second transplantation from complications of stage 2 GvHD of the gut and adenoviral sepsis. CG07 (CID) died 14 years post-transplantation while awaiting lung transplantation. All 3 patients who have died to date received their UCBT after 3.5 months of age and only CG07 had pretransplant infections. Two patients who received UCBT before 3.5 months of age (CG03, CG04) are alive and well to date. Five of the 8 patients who received their UCBT after the age of 3.5 months are alive and well. Five patients had infections pretransplant and 4 of these patients are alive and well to date.
Discussion
We present a retrospective outcome of a single center experience 10 years after UCBT in patients with SCID or CID. These immunodeficiency disorders are fatal if not diagnosed and treated in a timely manner. 10 HLA-matched sibling donor is the preferred source of stem cells for these patients; however, sibling donors are often not available. Prior studies demonstrate that results from HLA-matched allogenic UCB transplants were comparable with those from HLA-matched MUD transplants in children. 11
Hematopoietic progenitor cells in UCB have an expanded ability to proliferate, exceeding bone marrow. 7 UCB allows for a higher degree of HLA mismatch. UCB banks during the time of these transplants were limited, which reduced options for lifesaving treatments. Furthermore, the UCB unit can be collected at birth with little harm to the donor infant or mother, can be easily cryopreserved for decades, and can reconstitute lymphohematopoiesis in the transplanted recipient. 7 Other advantages are decreased likelihood of infections and immediate availability of HLA typed UCB. For SCID, UCBTs have higher rates of immune reconstitution and chimerisms than MMRD. 4 However, slow T cell recovery can contribute to increased morbidity and mortality.
The 10-year overall survival in our study was 80% (Fig. 5), comparable with survival data ranging between 56% and 71% reported in the literature.4,12–16 For our patients who survived beyond 2 months post-transplantation, myeloid engraftment was seen with median time of 10.5 days. The 2 patients who died had delayed myeloid engraftment with poor ANC recovery and/or no platelet recovery at the time of death, pointing to engraftment failure or delay as a contributing factor to overall mortality.
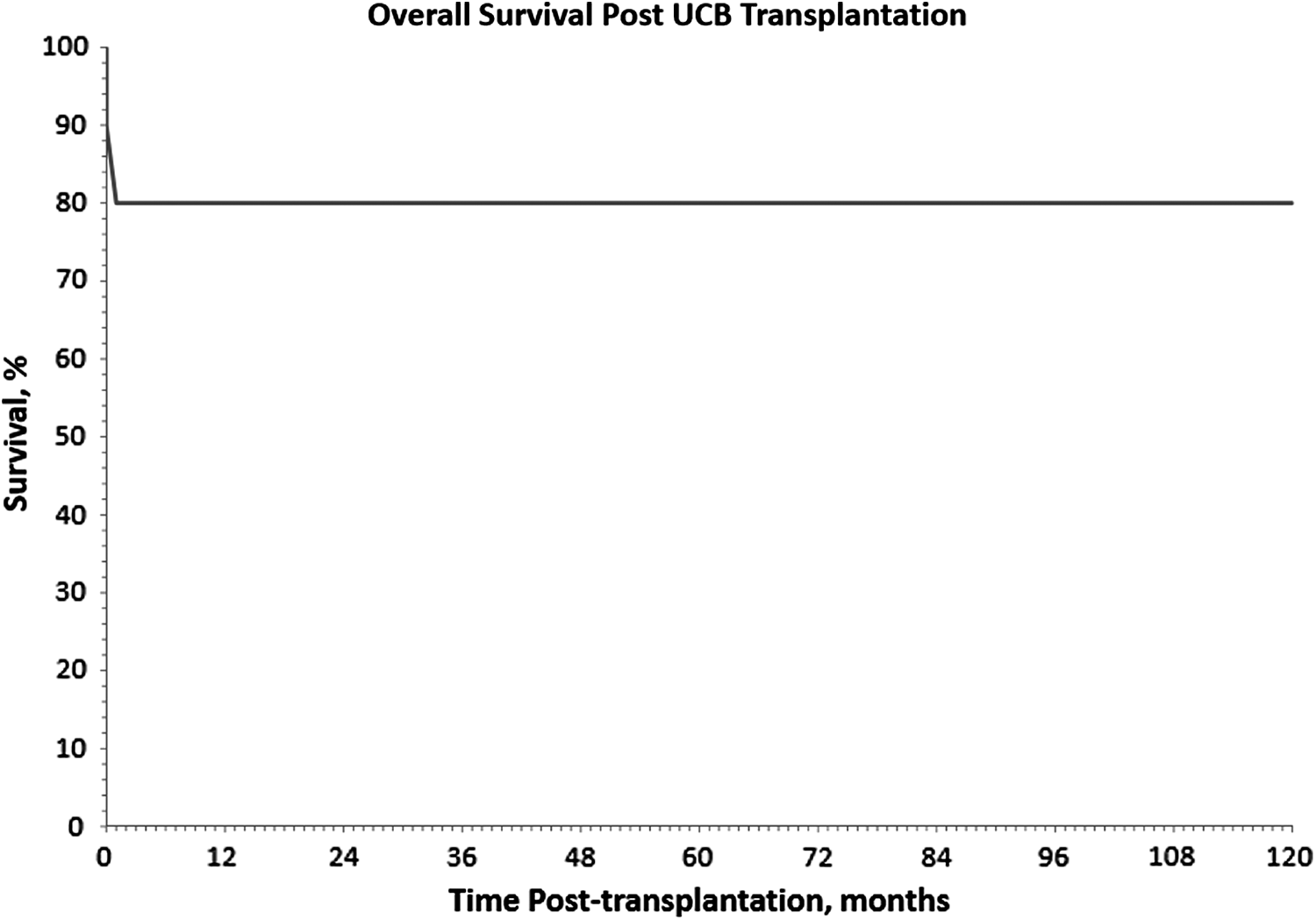
Overall survival post-UCBT.
After HSCT, slow T cell recovery can lead to complications with infections, significant morbidity and mortality, and worse overall prognosis during the first year post-transplantation.4,15 Engraftment of memory T cells is responsible for early appearance of T cell immunocompetence. Our patients' data demonstrate robust kinetics of immune reconstitution with an initial increase in the memory T cell population around 3–4 months post-transplant, which was followed by increase in naive T cells 12 months post-transplant. Our cohort showed a stable long-term T cell engraftment with normal lymphoproliferative responses to mitogens and alloantigens. Evaluation of B cell function in these patients demonstrated engraftment of donor B cells and normal tetanus and diphtheria toxoid antibody responses documented in 6 of 6 patients at 18–24 months post-transplantation.
In UCBT, a cell dose of 1 × 108 nucleated cells/kg of body weight typically resulted in engraftment time that is similar to peripheral blood stem cell transplant. 16 Our patients received total nucleated cell doses in the range of 6–16 × 108. There was no correlation between the dose and myeloid engraftment in our patients. Improved outcomes for patients with nonmalignant disease have been reported with higher doses, 11 which is associated with better survival rates. 7
Patients transplanted before 3.5 months old experience less pretransplant morbidity in terms of infections. Half of our patients had infectious complications pretransplant. However, the timing of transplant had a more profound impact on survival, when compared with the rate of infections. This is consistent with data previously reported,2,9,12 wherein survival rate was high regardless of donor type among infants who received transplants before 3.5 months old. Prior data demonstrate that prevention and successful treatment of infections are influential factors that determine a good transplantation outcome.
Ninety percent of our patients had grade 1 GvHD, which was mild and resolved with corticosteroids. In these patients, the high degree of HLA class 1 mismatch (3–6 antigen mismatch) did not predict the grade of GvHD. 14 This observation contrasts HLA-matched MUD transplantations, wherein there is a clear correlation between grade 4 GvHD and HLA disparity. In general, a higher disparity of HLA mismatch is associated with worse GvHD. However, the relative immaturity of the donor immune system allows for a greater degree of HLA mismatch in UCBT and this is likely one of the factors for decreased severity of GvHD in our series. 16 One of our patients (10%) developed grade 4 GvHD as compared with 25% in prior studies using UCB. 12 This patient had Rothmund Thomson syndrome, and it is possible that the underlying disease led to a higher grading of skin GvHD. Optimal GvHD prophylaxis for patients with immune deficiencies undergoing UCBT should be the focus of further research.
Autopsy findings from the patient who died from cardiac complications 19 days post-transplantation were suggestive of cardiac manifestations of cardiomyopathy, likely from cyclophosphamide toxicity leading to cardiac hemorrhage. 17 This patient had developed cardiac issues during conditioning and had no documented infections associated with myocarditis pretransplant. 17
Eighty percent of the patients underwent MAC with the remainder undergoing RIC. Prior studies have shown that up to 20% of patients with no prior conditioning require repeat conditioning therapy with HSCT or unconditioned boosts. 14 All our patients received conditioning regimens that may have contributed to a higher survival rate by allowing engraftment in the hematopoietic compartment. The timing of transplant took into account treatment of pre-existing infections, finding an appropriate UCB unit, and proceeding to transplant with a minimum delay after diagnosis. This may be a viable strategy to allow for cord blood expansion in the marrow space. Some authors report a lower risk of GvHD in patients who receive conditioning regimen pretransplant. 14 Also, the higher proportion of MAC regimens employed in this cohort may have contributed to higher frequency of complete donor chimerism observed, similar to prior reported studies.4,13,18
Conclusion
In conclusion, our study demonstrates that UCB is a viable approach to transplantation in SCID and CID patients when no sibling donor is available and results in durable immune reconstitution. Improvements in pre- and post-transplantation care, a better understanding of cell doses, improved HLA typing, and earlier detection of viral and fungal infections with more effective treatment options have improved outcomes in patients undergoing UCBT. Use of UCB as a stem cell source should be actively considered for patients with SCID or CID who lack a well-matched donor.
Footnotes
Author Disclosure Statement
No competing financial interests exist.