
Abstract
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare immune deficiency with a broad clinical presentation. IPEX syndrome causes dysfunctional regulatory T cells, increasing the risk of autoimmune diseases. In this case report, we describe a 7-year-old boy with lymphocytic interstitial pneumonia and bullous pemphigoid who was recently diagnosed with IPEX syndrome.
Introduction
Lymphocytic interstitial pneumonia (LIP) is characterized as a pulmonary lymphoproliferative disorder. It is classified as a diffuse pulmonary disease, often complicated by respiratory insufficiency due to increased lymphocytic infiltration of the alveolar septa, leading to traction bronchiectasis and pulmonary fibrosis. 1 The precise pathogenesis of LIP is unknown. However, LIP is considered to represent a nonspecific response to multiple stimuli. Among children, LIP can be associated with HIV and primary immunodeficiencies. Furthermore, associations with numerous autoimmune diseases suggest that LIP may represent an autoimmune condition. Accordingly, LIP is included in the provisional criteria for autoimmune-featured interstitial lung disease.1,2
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare immunodeficiency. Origin of IPEX syndrome is mutations of the transcription factor forkhead box P3 (FOXP3). Mutations in FOXP3 lead to dysfunction of regulatory T cells (Tregs), which is the main pathogenic event leading to autoimmunity in IPEX. The dysfunctional Tregs cannot control the development of T effector cells generating more T helper 2 cells, IL17-producing cells, and autoreactive B cells. The resulting autoimmune reaction increases the presence of inflammatory cells in the target organs and the production of autoantibodies. 3
The role of FOXP3 mutations in B cell development is not fully understood. However, patients may show altered antibody production with increased IgA and IgE.
Interestingly, patients can exhibit a variety of autoantibodies, some of which may be pathogenic. IPEX syndrome can be fatal if untreated.3,4
Autoimmune disorders have various clinical presentations; in particular, gastroenterological and dermatological symptoms are often described in conjunction with IPEX syndrome.5,6
Bullous pemphigoid (BP) is an autoimmune blistering skin disease most often seen in the elderly population; the disease is extremely rare among children. 7
We present a now 7-year-old boy with both LIP and BP suffering from IPEX syndrome.
Case
The boy was born at term as the second child of healthy nonconsanguineous parents with no family history of primary immunodeficiency or autoimmune disorders. At the age of 1 year, he was admitted to hospital due to failure to thrive with a drop of 2 standard deviations (SDs) in 3 months (+1 to −1 SD) and hypoxia (saturation 70%–80% without oxygen therapy). High-resolution computed tomography of the lungs revealed diffuse ground glass opacities and focal consolidation. Owing to the findings of chronic pulmonary changes, a meticulous examination was initiated including bronchoscopy, bronchoalveolar lavage, brush biopsy, and a lung biopsy. The bronchoscopy showed inflamed mucosa and edema and the brush biopsy showed unspecific inflammation dominated by lymphocytes and neutrophils. Staphylococcus aureus and Hemophilus influenzae species were identified by the bronchoalveolar lavage. Blood tests showed intermittent eosinophilia and selective IgA deficiency. Antinuclear antibodies (ratio 2.4) and antidouble-stranded DNA antibodies (13 × 10^3 IU/L) were slightly increased. The boy did not have any endocrinological manifestations.
Histopathology of a surgical lung biopsy showed marked expansion of the alveolar interstitium by a cellular infiltrate comprising predominantly lymphocytes and histiocytes mixed with scattered neutrophils and eosinophils without atypical cells or signs of malignancy. The pulmonary vasculature was normal (Fig. 1). The diagnosis of LIP 8 was established after a second opinion on the biopsy results at Royal Brompton Hospital in London.
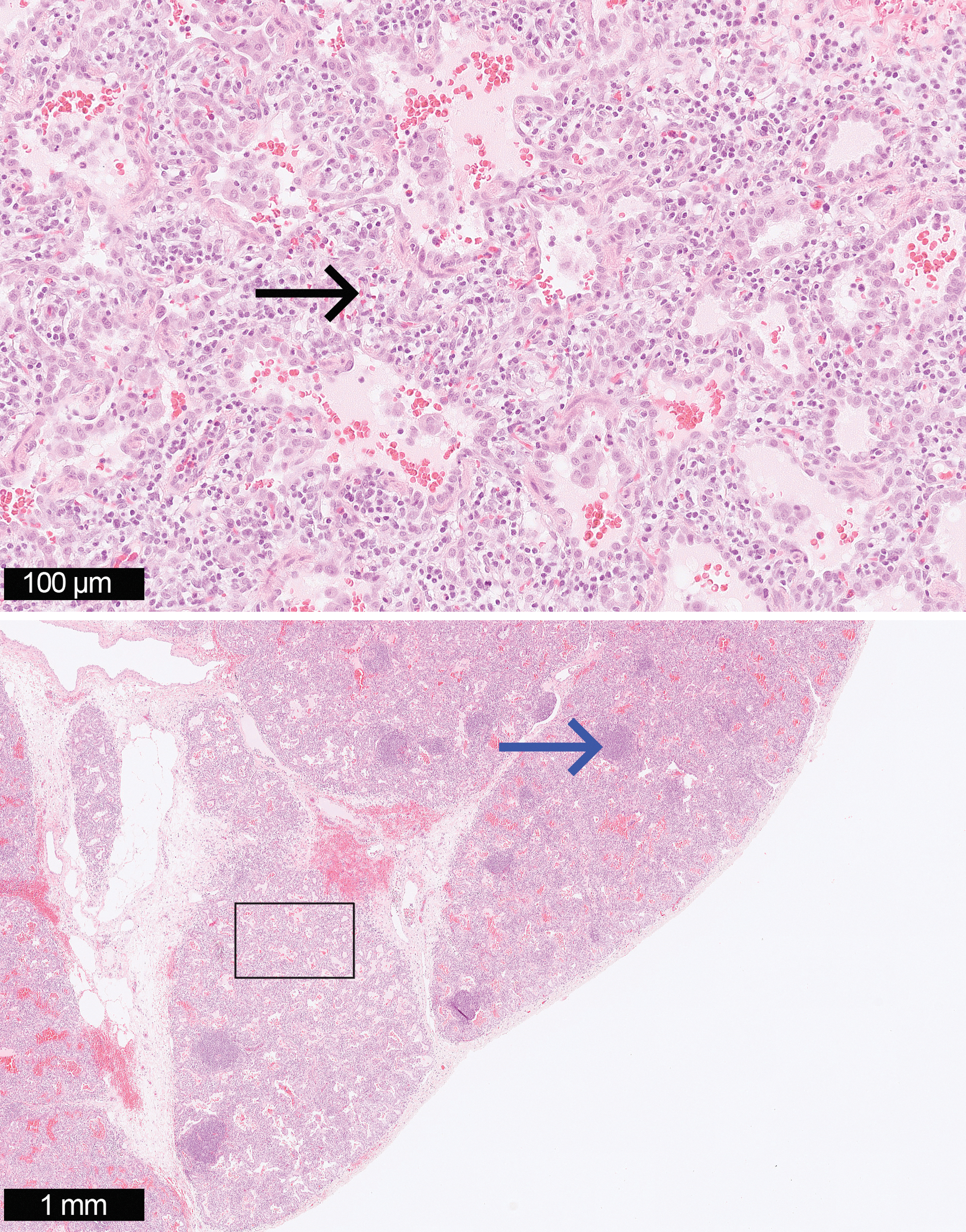
Microscopy of surgical lung biopsy showed marked expansion of the alveolar interstitium by a cellular infiltrate comprising lymphocytes (black arrow) with scattered germinal centers (blue arrow) adjacent to small airways (H&E × 25 and × 200). H&E, hematoxylin and eosin. Color images are available online.
The patient's LIP was treated effectively with high-dose pulse methylprednisolone 25 mg/kg intravenously (IV) daily for 3 days per month for 6 months with systemic prednisolone 2 mg/(kg·day) in between IV treatment. Later, due to frequent bacterial lung infections, prophylactic therapy with azithromycin (5 mg/kg every second day) was initiated. Daily dose of oral prednisolone (5 mg twice a day) was tapered and regulated over several months; treatment was discontinued when the patient was ∼3.5 years old. Prophylactic therapy with azithromycin continued, thus minimizing secondary infections and pulmonary exacerbation.
The patient developed multiple psychological side effects to prednisolone treatment. However, azithromycin was well tolerated.
Nonspecific eczematous skin symptoms developed when the boy was ∼2 years old. Skin biopsies indicated dermatitis. Around 4 years of age, the skin symptoms progressed into a severe blistering disease mainly affecting the face, the gluteal region, arms, and legs (Fig. 2A, B). New skin biopsies showed inflammation with eosinophilia. Histopathology and both direct and indirect immunofluorescence on salt-split skin biopsies showed positive reactions with an IgG roof pattern. Furthermore, blood tests demonstrated anti-BP180 autoantibody, which is a significant autoantibody in the pathogenesis of BP. 9

The skin disease was resistant to topical treatment with both potent corticosteroids and calcineurin inhibitors. Methotrexate, which often is considered first-line treatment of BP, was not initiated due to a precautionary principle. 10 Immunomodulatory treatment with intravenous immunoglobulins for 6 months showed no effect. Prednisolone caused psychological side effects, dapsone lead to increased dyspnea, azathioprine to gastrointestinal side effects, and coated mycophenolate mofetil sodium showed no effect. Most recently, cyclosporine A was initiated with some effect on the skin and with tolerable side effects. The child is followed regularly in the outpatient clinic.
Based on the complicated course of both lung and skin diseases, a laboratory evaluation of primary immunodeficiency was performed. Flow cytometry of peripheral lymphocytes revealed a decreased fraction of Tregs (CD4+CD25+CD127-FoxP3+) at 2.9% of total CD4+ T cells (Fig. 2C–G) (reference 95% interval: 4.0%–10% of CD4+ T cells). The remaining immune phenotyping data, including other T cell subpopulations, B cells, and NK cells, were normal (Table 1). Whole exome sequencing revealed a hemizygous FOXP3 variant (c.1150G>A; p.A284T), which was confirmed by Sanger sequencing. This variant is predicted to be deleterious by a high combined annotation-dependent depletion score of 25 and has previously been reported to cause IPEX. 11 The variant is associated with significantly impaired suppressive function of Tregs and is classified as “disease causing” in the human genetic mutation database. Biparental sequencing revealed a de novo event in the index patient.
Immune Phenotyping Data
Cells per μL peripheral whole blood determined by flow cytometry. Reference interval: Shearer WT et al., JACI 2003 (PMID: 14610491).
This specific IPEX variant has previously been associated with varying fractions of Tregs, a heterogeneous clinical disease severity and significantly impaired suppressive function of Tregs. 11
Parental permission was granted for use of patient photograph and case presentation.
Discussion
LIP and BP are both rare in children.7,12 In this case, thorough examination revealed a specific FOXP3 mutation, which is a known cause of IPEX syndrome.
Autoimmune enteropathy is a hallmark of IPEX syndrome. Gastrointestinal symptoms may vary in severity from neonatal severe diarrhea to symptoms mimicking inflammatory bowel disease.3,13 In this patient, absence of gastrointestinal symptoms apart from mild gastritis was a complicating factor in the diagnostic procedure.
Other possible differential diagnoses of LIP are coatomer protein complex subunit alpha syndrome (COBA), CD25 deficiency, CD122 deficiency, and lipopolysaccharide responsive and beige-like anchor protein (LRBA) deficiency. 12
Skin involvement is common in IPEX syndrome, most frequently dermatitis and allergic reactions, but pemphigus nodularis has been described in 1 case. 5 The essential role of Tregs in the development of BP has recently been reported by Muramatsu et al. who also identified autoantibodies against BP180 and BP230 in patients with IPEX syndrome. 14 LIP is known to be associated with autoimmune and immunodeficiency disorders. 2 Nevertheless, LIP as a clinical manifestation of IPEX syndrome is rare.15,16
Conclusion
The pathogenesis of LIP is still poorly understood. A diagnosis of LIP must always initiate a thorough examination to reveal any possible underlying cause. Furthermore, in cases of severe treatment-resistant BP, a meticulous evaluation of the patient is advisable. In patients who develop multiple autoimmune diseases, the possibility of an underlying primary immunodeficiency should be investigated.
Footnotes
Acknowledgment
We appreciate the effort of Line Bille Madsen from the Department of Pathology, Aarhus University Hospital, for sharing her expertise regarding histology pictures and description.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors received no financial support for the research, authorship and publication of this article.