
Abstract
Background:
RAS guanyl-releasing protein 1 (RASGRP1) deficiency is characterized by immune dysregulation and Epstein–Barr virus (EBV)-related lymphoproliferation. Diffuse mesangial sclerosis is one of the infrequent causes of infantile nephrotic syndrome.
Case Presentation:
Here, we described a 7-year-old girl who was diagnosed with diffuse mesangial sclerosis at 5 months old and subsequently developed chronic bilateral neck swelling at the age of 3 years. Clinical assessment and investigations revealed a complex clinical picture, including recurrent cervical lymphadenopathy and recurrent infections. Further evaluation revealed immunological deficiencies, autoimmune lymphoproliferative syndrome-like illness, chronic EBV infection, and ultimately Hodgkin lymphoma. Genetic testing identified a RASGRP1 homozygous loss-of-function variant with both parents being carriers.
Conclusion:
This is the first reported case of RASGRP1 deficiency in Malaysia, and we highlight the challenges clinicians face when the disease manifests in varied presentations.
Introduction
RAS guanyl-releasing protein 1 (RASGRP1) is a guanine-nucleotide-exchange factor and activator of the RAS-MAPK (Ras/mitogen-activated protein kinase) pathway following T-cell receptor signaling. 1 This protein is highly expressed in T-cells, and to a lesser extent in B-cells and natural killer (NK) cells. 2 Several studies have demonstrated that RASGRP1 is essential for T-cell development, homeostasis and differentiation, and therefore RASGRP1 deficiency leads to impairment of T-cell function including activation and proliferation.3,4 Loss-of-function variants in the RASGRP1 gene have been identified in patients with early onset primary immunodeficiency and a predisposition to Epstein–Barr virus (EBV)-driven lymphoproliferative disease.5–7 Autoimmune association has also been reported as a result of defective RASGRP1 function.3,7
Diffuse mesangial sclerosis (DMS) is an infrequent cause of childhood nephrotic syndrome. This condition is characterized by progressive sclerosis of the mesangial matrix with minimal or absent mesangial cell proliferation. 8 While some cases of DMS may have genetic or hereditary factors, the exact cause of this condition remains elusive. 9 There is currently no substantial evidence supporting a role of immune dysregulation in DMS. On the other hand, RASGRP1 variants are not typically associated with renal conditions, further emphasizing the complex presentation of RASGRP1 deficiency.
Given the intricate nature of RASGRP1 variants with immune deficiencies, autoimmunity, EBV-driven lymphoma, and the uncommon occurrence of DMS-related nephrotic syndrome in this patient, we describe this unique case aiming to shed light on the complex clinical course, immunological findings, and genetic diagnosis.
Case Illustration
In 2020, a 5-year-old girl was referred to our clinical immunology unit for chronic bilateral neck lymphadenopathy with suspected primary immunodeficiency (Fig. 1A). She was the second of five siblings, born at term to consanguineous Malay parents who were first-degree cousins (Fig. 1B
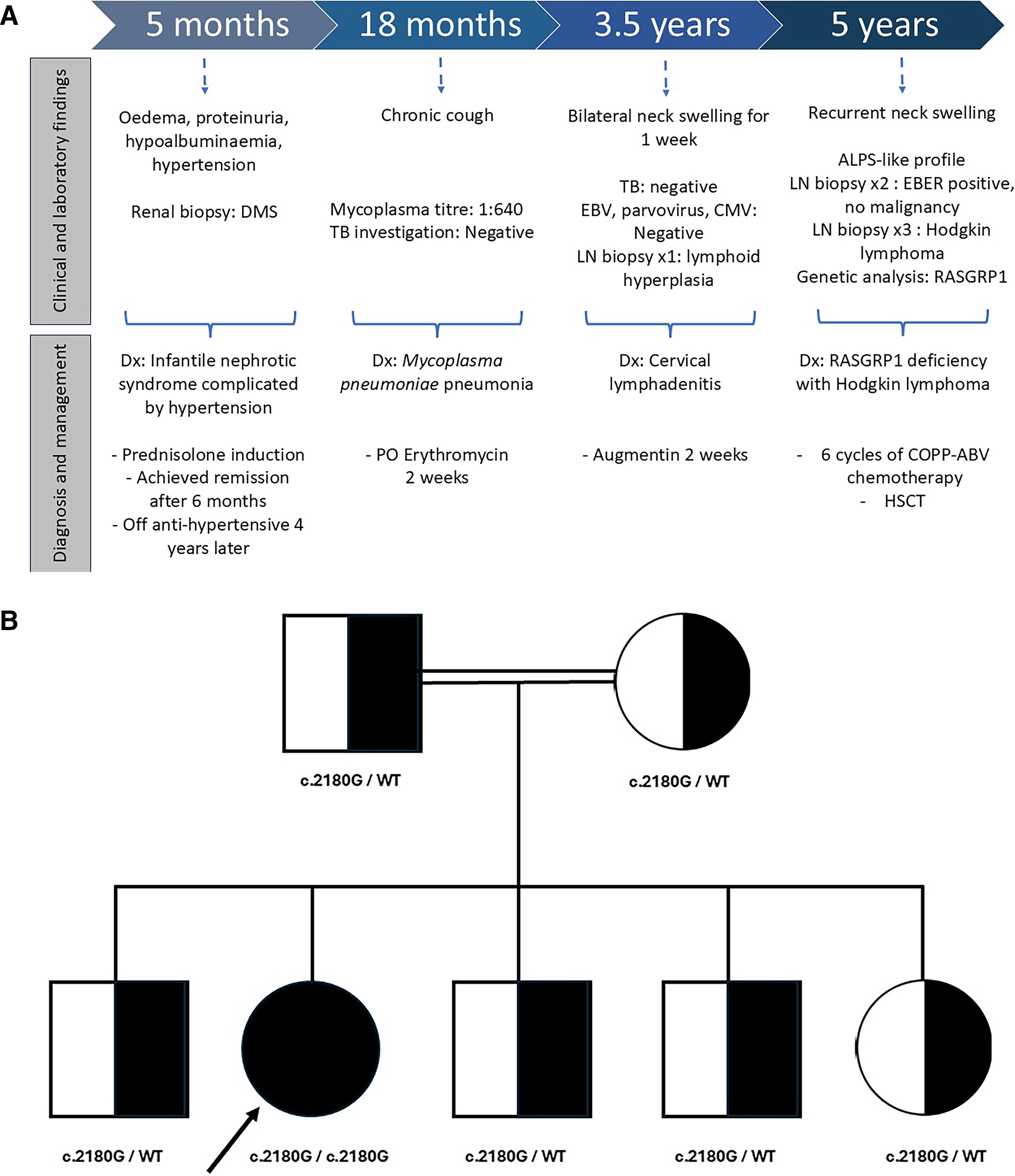
She was healthy until the age of 5 months, when she developed generalized edema, proteinuria, and hypoalbuminemia and later was diagnosed with infantile nephrotic syndrome. The disease was complicated by hypertension requiring triple antihypertensive (nifedipine, spironolactone, and enalapril). A comprehensive investigation including screening for toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus (TORCHES); human immunodeficiency virus (HIV); and autoimmune screening yielded negative findings. She initially displayed a steroid-resistant type and had a renal biopsy done at age 8 months, which revealed diffuse mesangial sclerosis. She subsequently responded to prednisolone and achieved remission within the next 6 months. During clinic follow-ups, she was found to have asymptomatic transaminitis, which resolved spontaneously at age 18 months. The anti-hypertensive medications were tapered down and discontinued when she reached 4 years old.
She had mycoplasma pneumonia at the age of 18 months when she presented with a prolonged cough for 3 weeks. The mycoplasma titer was 1:640, and tuberculosis (TB) workup was negative. She was treated with erythromycin and discharged well. Ever since then, she had intermittent bronchospasm episodes with interval symptoms. Hence, she was diagnosed with mild persistent bronchial asthma.
At 3.5 years old, she presented again with a 1-week history of left neck swelling of 3 × 3 cm in size. There were no constitutional or respiratory symptoms. Initial investigations for TB and viral causes, including EBV, parvovirus, and cytomegalovirus (CMV), yielded negative results. A neck ultrasound showed bilateral lymphadenopathy. Due to a lack of improvement with adequate antibiotics, an excisional lymph node biopsy was done. The histopathology examination reported reactive lymphoid hyperplasia, unlikely to be lymphoma.
Despite initial improvement, the cervical lymphadenopathy returned 6 months post-biopsy, involving both sides and gradually increasing in size over the next 2 years. Computed tomography (CT) of neck and thorax revealed multiple cervical and mediastinal lymphadenopathy with multiple nodules in the right lower lobe and total collapse of the right middle and left lower lobes secondary to compression of the surrounding nodes. Along the course of illness, she had several episodes of pneumonia and chronic suppurative otitis media as well as failure to thrive. Summary of the patient’s key clinical characteristics is shown in Table 1.
Summary of Key Clinical Characteristics of the Patient
EBV, Epstein–Barr virus.
On examination, she was small for her age; weight 16.4 kg (below 5th centile) and height 108 cm (below 5th centile). Vital signs were stable. There were bilateral multiple cervical lymphadenopathies measuring 10 × 5 cm and 12 × 7 cm over the right and left sides, respectively. They were non-tender, firm, mobile, and some were matted together, with no overlying skin changes (Fig. 2A and B). She had a palpable liver measuring 4 cm below the right costal margin with palpable tip of spleen. Bilateral otoscopy examination showed abundant pus discharge, and the tympanic membranes were unable to be visualized. Other systemic examinations were unremarkable.

A series of comprehensive laboratory tests were carried out. Serial complete blood count did not show any signs of cytopenia. Her absolute numbers of neutrophils and lymphocytes were normal. The inflammatory markers showed erythrocyte sedimentation rate (ESR) 76 mm/hour and C-reactive protein (CRP) 26 mg/L. Multiple TB workups, including TB culture and TB polymerase chain reaction (PCR), were negative. Both EBV immunoglobulin M and EBV PCR were positive, and her EBV viral load remained detectable but consistently below 1000 copies (range: 351–891 copies/mL).
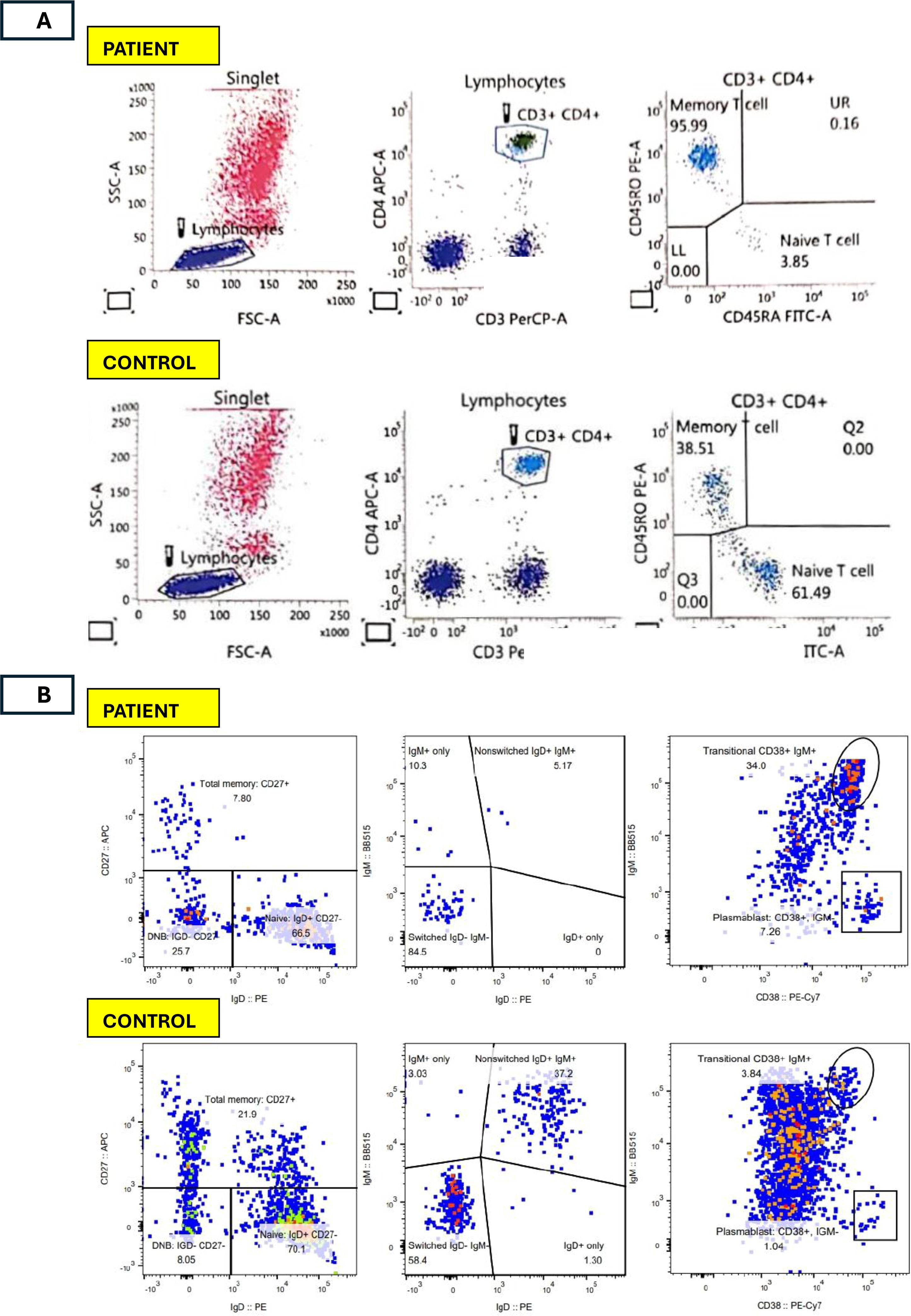
Testing for other viruses and serial blood cultures were again negative. Autoimmune workup revealed a positive direct Coombs test, antinuclear antibody (ANA) with a speckled and homogenous pattern at a titer of 1:320, and positive anti-double-stranded DNA (anti-dsDNA) antibodies. Vitamin B12 was 1303 pmol/L (normal range 156–672 pmol/L). Immunological data revealed low T cells and B cells, low CD4+ and CD8+ T cells, high NK cells, hypergammaglobulinemia, low class-switched memory B cells, low naïve T cells, elevated C3 and normal C4. Alpha/beta double-negative T-cells (DNT cells) count was inconclusive. With the evidence of autoimmunity and lymphoproliferation, clinical autoimmune lymphoproliferative syndrome (ALPS)-like was suspected. Flow cytometry figures reflecting the relevant immunological profiles are shown in Figure 3A, B, and C.
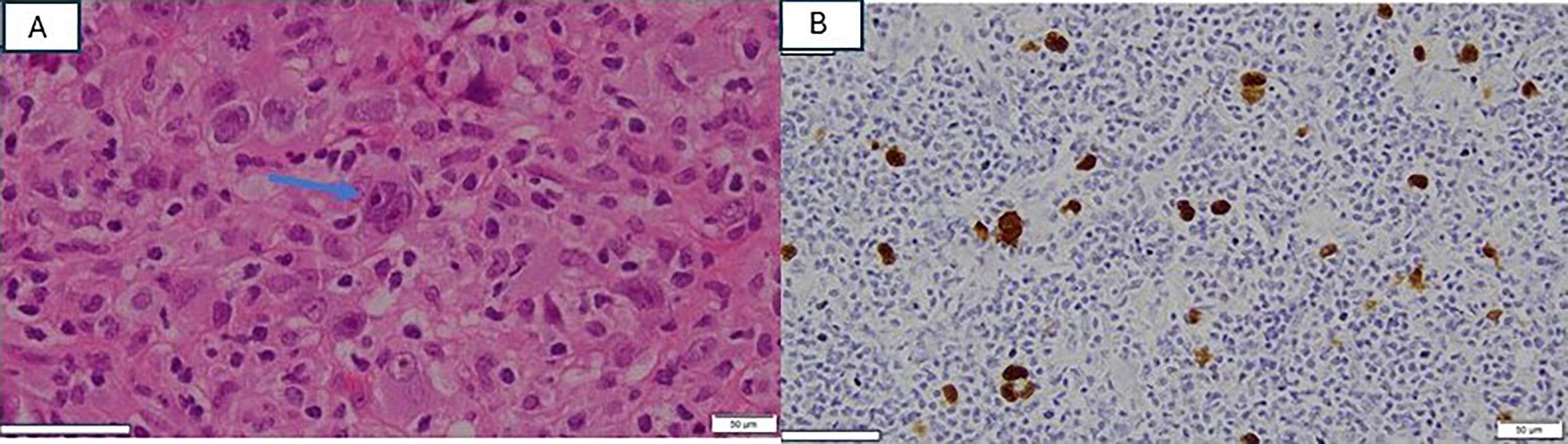
Throughout the course of her illness, she underwent three cervical lymph node biopsies. The second biopsy revealed EBV lymphoproliferative disease without evidence of malignancy. Despite the continuous progression of lymphadenopathy, we were not able to proceed with a follow-up lymph node biopsy as the hospital was converted into a COVID-19 hospital and elective surgical procedures were suspended. Eventually, the third excisional biopsy performed in 2021 (Fig. 2C) revealed classic Hodgkin lymphoma; nodular sclerosis subtype with scattered Reed–Sternberg cells was present on histopathology examination (Fig. 4A). Immunohistochemical staining of the neoplastic cells was positive for CD30, and weak positive for CD20, CD79, and PAX5. In situ hybridization for EBV-encoded RNA was positive in the majority of atypical lymphoid cells (Fig. 4B). The genetic test for 407 variants related to primary immunodeficiency revealed a homozygous loss-of-function pathogenic variant in exon 16 of RASGRP1 gene (NM_005739.4; c.2180G>A, p.Trp727*). Both parents and other siblings were found to be heterozygous carriers in line with autosomal recessive inheritance (FIG. 5).
She was initially started on mTOR (mechanistic target of rapamycin) inhibitor for the possible diagnosis of ALPS-like, and the lymphadenopathy showed a temporary reduction in size. After the diagnosis of Hodgkin lymphoma, she was commenced on six cycles of Cyclophosphamide, Oncovin (Vincristine), Prednisone, Procarbazine – Adriamycin (Doxorubicin), Bleomycin, Vinblastine (COPP-ABV) chemotherapy. However, a positron emission tomography CT study after completion of chemotherapy showed active avid fluorodeoxyglucose (FDG) uptake at the cervical lymph node area suggestive of progression of disease. Her repeated blood EBV PCR showed 10610 copies/mL. Intravenous rituximab was given to overcome progressive EBV viraemia. She successfully underwent hematopoietic stem cell transplant (HSCT) from her human leukocyte antigen (HLA)-matched younger sister. The conditioning regimen included Fludarabine (160 mg/m2) from Day −6 to Day −4, followed by Melphalan (70 mg/m2) from Day −3 to Day −2, as part of the pre-transplant protocol (Fig. 6). She is currently 10 months post-HSCT with a stable graft and achieved continuous remission for both EBV viremia and lymphoma. Table 2 illustrates the immunological profiles before and after bone marrow transplant.
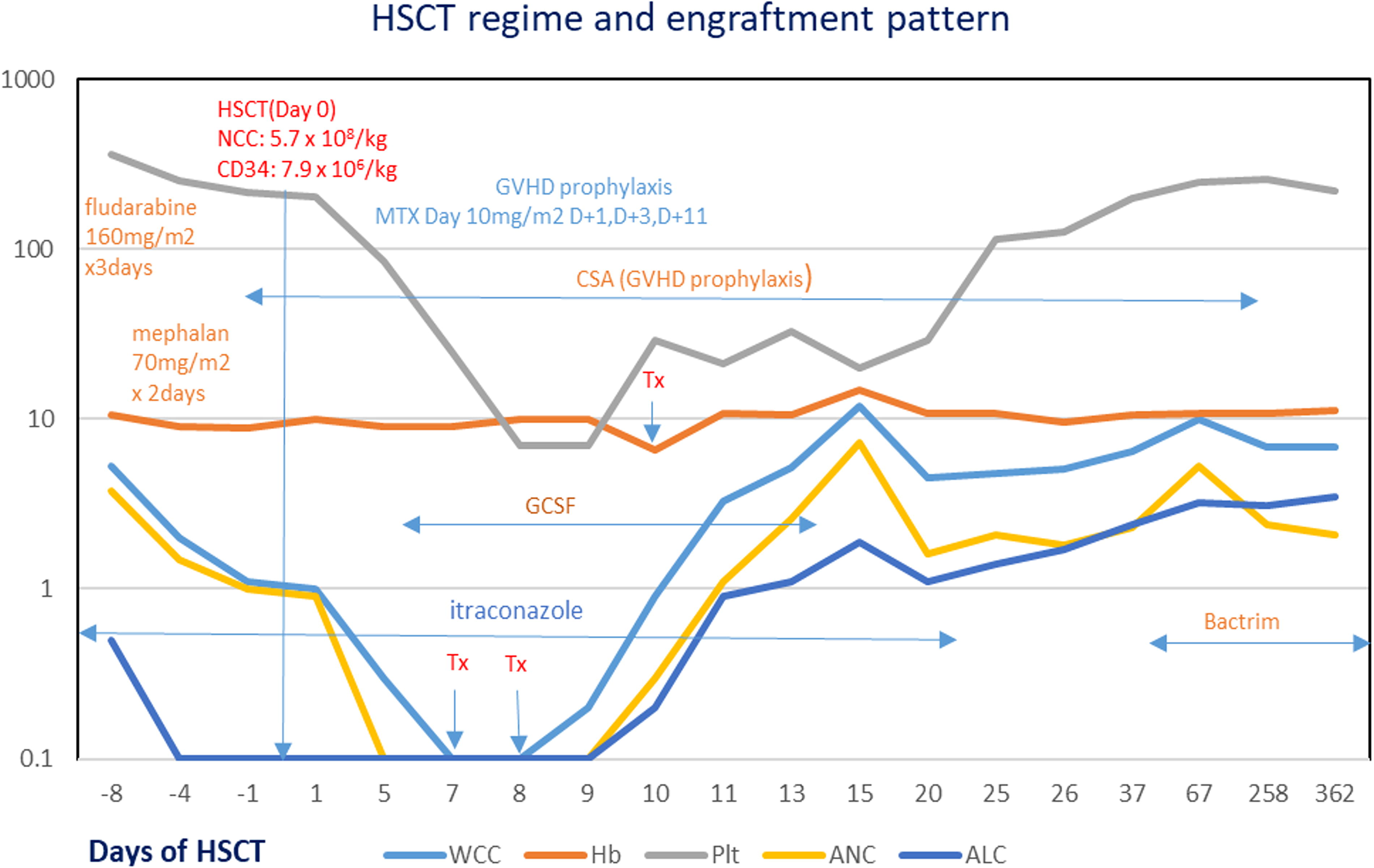
Timeline of hematopoietic stem cell transplantation (HSCT).
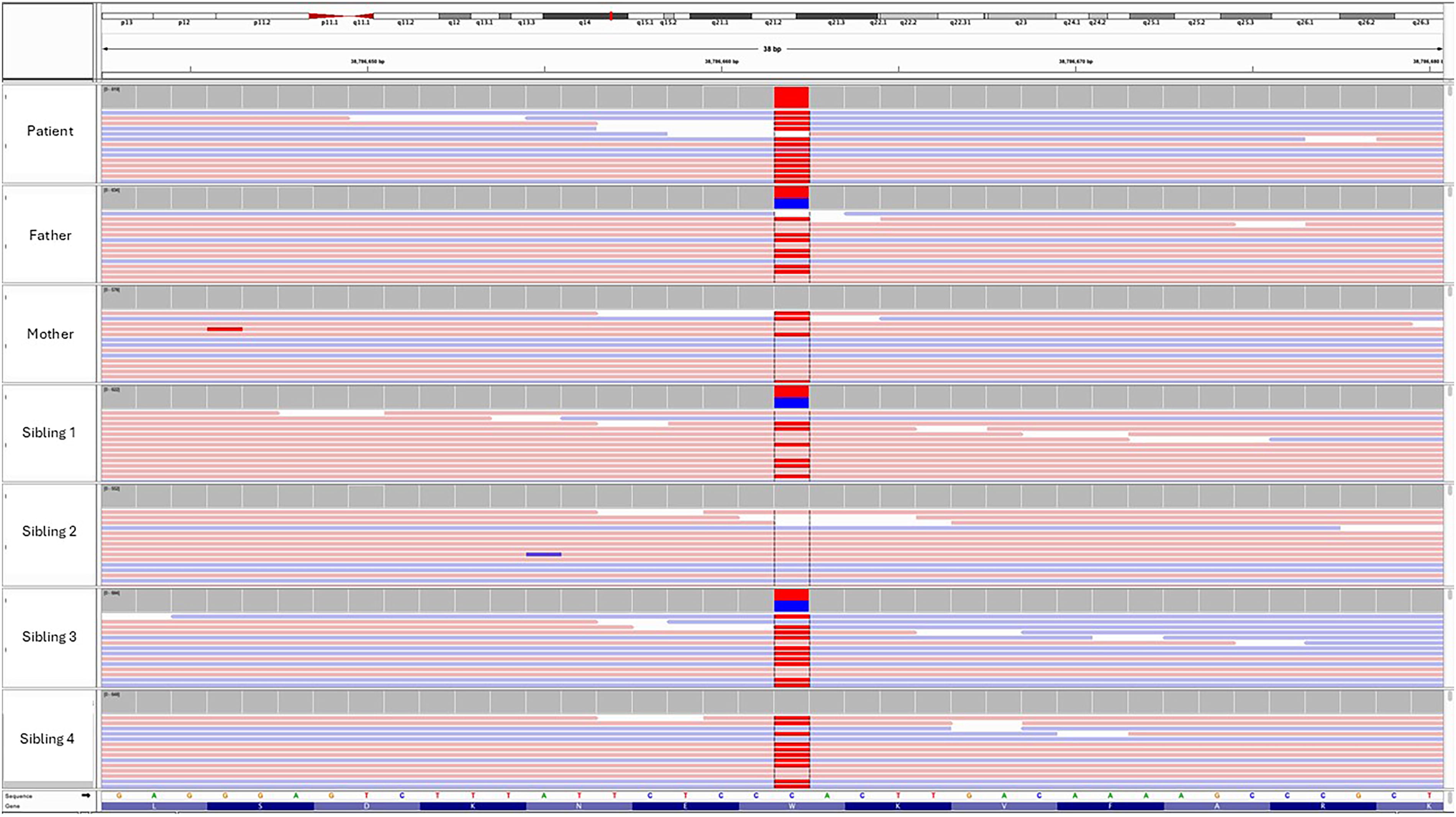
Integrative Genomics Viewer (IGV) visualizations of Next-generation sequencing (NGS) data for the RASGRP1 gene in the patient and their family members. The red box highlights the pathogenic variant detected at position c.2180G>A (p.Trp727*), observed in both alleles in the patient (homozygous), indicating a loss-of-function mutation.
Immunological Profiles of the Patient Pre- and Post-HSCT
DNT cells, double-negative T-cells; HSCT, hematopoietic stem cell transplant; IgA, Immunoglobulin A; IgD, Immunoglobulin D; IgE, Immunoglobulin G; IgE, Immunoglobulin G; IgM, Immunoglobulin M; NK, natural killer; TCR, T-cell receptor; PHA, phytohemagglutinin.
Discussion
In animal studies, RASGRP1-deficient mice demonstrated compromised immune responses and impaired pathogen-specific T-cell reactions, leading to delayed pathogen clearance upon exposure to bacteria and viruses. 10 It is also important in the cytokine production and cytotoxicity of natural killer cells. 11 In humans, RASGRP1 deficiency was first described in 2016 by Salzer et al. 4 Subsequently, nine more cases have been reported in the literature. The main clinical features observed include recurrent infections, hepatosplenomegaly, lymphadenopathy, EBV-associated lymphoproliferation, B cell lymphoma, and autoimmune manifestations,3–7 mirroring the presentation observed in our patient, except for the distinctive presence of infantile nephrotic syndrome. RASGRP1 deficiency has been linked to a reduction in NK cytotoxic activity, defective B cell formation, proliferation, and signaling, impaired CD8 signaling and proliferation, and CD4 T cell lymphopenia.5,4 Furthermore, RASGRP1 deficiency is recognized for its autoimmune associations, with previous literature reporting autoimmune manifestations ins 5 out of 8 patients. 6 Our patient displayed low T cell, B cell, and NK cell counts, along with evidence of hypergammaglobulinemia, and positive autoimmune markers. Flow cytometry of peripheral blood showed increased DNT cells of the CD3+ T cell population, but normal DNT cells of the total lymphocytes, which does not support a definite diagnosis of ALPS. However, the clinical criteria for ALPS-like conditions are present which are characterized by immune dysregulation and defined by the co-occurrence of autoimmunity and lymphoproliferation. 13 It is important to note that there are currently no established clinical or laboratory guidelines specifically for diagnosing ALPS-like patients. Molnar et al., found that only 51.5% of probable ALPS-like patients presented with abnormally high DNT cell counts, indicating that this characteristic is not pathognomonic for this type of syndrome. 14
Apart from its role in immune defense, RAS signaling has been associated with malignant transformation. RASGRP1-deficient T cells also decrease CD27-dependent proliferation toward CD70-expressing EBV-transformed B cells, which is essential for the expansion of antigen-specific T cells during EBV infection. 7 This impairment in activation-induced cell death heightens susceptibility to EBV infection, contributing to the development of autoimmune lymphoproliferative syndrome (ALPS)-like illness and predisposing individuals to Hodgkin lymphoma. The reported outcomes of EBV-driven lymphoma in people with RASGRP1 deficiency vary. A case reported by Platt et al., in which the patient died of lymphoma-related complications after receiving chemotherapy, emphasizes the importance of prompt HSCT in RASGRP1-deficient patients. 5 Subsequent cases, as described by Somekh et al, have demonstrated more favorable outcomes with HSCT, demonstrating complete remission, as observed in our patient. 6
In addition to the predominant clinical features observed in previously reported cases, our patient was presented with a distinctive history of DMS infantile nephrotic syndrome. DMS often presents with nephrotic syndrome at birth or within the first year of life. 15 It is characterized by progressive sclerosis of the mesangial matrix with minimal or absent mesangial cell proliferation, hypertrophy of podocytes in early disease thickened basement membranes, diminished patency of capillary lumen and, crown-like appearance of vacuolated podocytes in advanced disease. 8 While DMS is commonly associated with genetic syndromes, it can occasionally manifest in an isolated form. Notably, DMS has also been observed in infantile nephrotic syndrome caused by infections such as CMV and toxoplasmosis. 2 In our patient’s case, upon presenting with nephrotic syndrome, various infective causes were ruled out, including TORCHES infections, HIV, and viral hepatitis.
As far as we know, there is no evidence yet to link RASGRP1 pathogenic variants to nephrotic syndrome. RASGRP1 pathogenic variants are typically associated with immune dysregulations rather than renal diseases. However, it is important to note that individual cases and genetic variations can present with unique manifestations. In our case, the RASGRP1 pathogenic variant creates a premature translational stop signal (p.Trp727*) in the penultimate exon (NM_005739.3, exon 16), which is expected to result in an absent protein due to nonsense-mediated mRNA decay or a disrupted protein product. Loss-of-function variants in RASGRP1 are known to be pathogenic (PMID: 11017103, 27776107, 28822832). In contrast, our patient could have a second genetic diagnosis as an underlying etiology causing the DMS nephrotic syndrome. Unfortunately, we were not able to send another genetic testing for the kidney panel because of family’s financial constraint.
One study found a novel association of RGS1 and RASGRP1 variants in immunoglobulin A nephropathy indicating that RASGRP1 variants are likely to influence disease through mechanisms regulating gene expression. 16 Yang et al. revealed a potential association between EBV infection and renal damage, characterized by varying degrees of hematuria or proteinuria, as well as edema and hypertension. However, it should be noted that the proportion of patients who underwent renal biopsy in that study was low. Interestingly, they also found that pediatric patients with EBV infection who exhibited renal damage had notably lower blood CD4+/CD8+ T cell ratios compared to the non-renal damage group. 17 This observation suggests a possible link between the development of renal damage and the suppression of cellular immune function following acute EBV infection. Unfortunately, our patient was not specifically tested for EBV infection at the time of presentation with nephrotic syndrome. Hence, it remains uncertain whether EBV could have been a contributing factor in her case, underscoring the need for further investigation into this potential mechanism. Therefore, in rare instances, it is conceivable that RASGRP1 variants could affect the inflammatory and regulation of the renal system, but further research and clinical observations are needed to establish a definitive association.
Conclusion
In conclusion, this unusual presentation of RASGRP1 deficiency with infantile nephrotic syndrome and EBV-induced Hodgkin lymphoma may shed new light to exploit the potential pathogenesis for those two diseases. The underlying link between these two conditions necessitates additional research to gain a comprehensive understanding of the association, which may help us better understand and improve the care and outcomes of these individuals in the future.
Footnotes
Acknowledgment
The authors extend their gratitude to the patient and her family for their invaluable participation in this study.
Informed Consent
Written informed consent for the publication of the patient’s medical details and accompanying images was obtained from the patient’s father.
Authors’ Contributions
Conceptualizations: K.N.M.N. and I.H.I. Data Collection: S.E.N., H.R., K.N.M.N., and M.A.Z.A. Analysis and interpretation: H.A., V.J., C.Y-R.H. Writing: Original Draft: K.N.M.N. Writing Review and Editing: I.H.I. Supervision: A.F.Z., I.S.O., and M.N.M.U. Critical Revision: M.A.Z.A. and I.H.I. Final Approval: All authors.
Permission to Reproduce
This case report presents an original clinical observation conducted by the authors. The findings and insights provided in this report are not a reproduction of any previously published work. The study aims to contribute novel information to the existing literature, shedding light on RASGRP1 deficiency in the context of its associated clinical manifestations. The authors declare that this case report has not been submitted for publication elsewhere.
Compliance with Ethical Standards
This case report was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki. All procedures were carried out with informed consent from the patient’s legal guardians and assent from the patient’s first-degree relatives, in line with the ethical standards of the institution. Informed consent was obtained for the use of medical data and images in this report, ensuring that patient anonymity and confidentiality were strictly maintained. The patient’s identity remains confidential, and all identifying information has been omitted from the article to protect privacy.
Data Availability Statement
All data underlying the results are available as part of the article and no additional source data are required.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding Information
This report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.