
Abstract
Introduction:
Only a few Lactobacillus casei phages have so far been characterized. As several L. casei strains are part of probiotic formulations, bacteriophage outbreaks targeting these strains can lead to critical losses within the dairy industry.
Materials and Methods:
A new L. casei phage was isolated from raw milk obtained from a milking yard from the province of Buenos Aires. The phage genome was sequenced, annotated, and analyzed. Morphology was determined by electron microscopy and the host range was established.
Results:
Lactobacillus phage vB_LcaM_Lbab1 is a member of the Herelleviridae family and features a host range including L. casei/Lactobacillus paracasei and Lactobacillus kefiri strains. We further analyzed the baseplate proteins in silico and found putative carbohydrate binding modules that are responsible for host recognition in other Lactobacillus phages.
Conclusions:
A new Lactobacillus phage was isolated and characterized. The focus was made on its host recognition mechanism, pointing toward the development of future strategies to avoid deleterious infections in the dairy industry.
Introduction
L
In the context of an international phage course at the University of Buenos Aires, directed toward graduate students and young researchers from Latin America, several phages were isolated, sequenced, and annotated using L. casei strains as bacterial hosts (see commentary in this issue). In this study we present the characterization of one of those phages, isolated from raw milk, obtained from a milking yard from the province of Buenos Aires in Argentina.
Materials and Methods
Bacterial strains and culture conditions
L. casei BL23 and L. casei subsp. casei ATCC 27139 were used as isolation hosts and were cultured in De Man, Rogosa and Sharpe (MRS) at 37°C without agitation. Solid culture was performed in MRS (De Man, Rogosa and Sharpe) agar. Lactobacillus paracasei subsp. paracasei ATCC 27092, L. acidophilus ATCC 4356, and Lactobacillus kefiri JCM 5818 were used for host range assays and were cultured using the same conditions as the isolation hosts.
Isolation, amplification, and stock preparation
Lactobacillus phage Lbab1 was isolated from a sample of unpasteurized milk from milking yards of the local dairy industry. In brief, to 5 mL of milk a few drops of 1 N NaOH were added until curd formation. Samples were centrifuged for 10 min at 4000 g and the supernatant was filtered using a 0.22 μm membrane. After enrichment in MRS medium overnight at 37°C using L. casei BL23 as host, isolation and purification were carried out through the double-layer assay as described by Kropinski et al. 7
Host range assay
The host range assay was performed using a double-layer assay as previously described 7 ; however, incubation of the plates was performed for 24 h at 30°C in the presence of 10 mM CaCl2.
The high titer stocks were prepared using the double-layer method. In brief, 80 Petri dishes were incubated with an MRS top agar layer containing 100 μL of log-phase L. casei BL23 and 100 μL of a phage suspension containing ∼500–1000 PFU. This provided confluence of plaques but not complete lysis after incubation for 24 h at 30°C. The plates were covered with buffer for 24 h at 4°C. The overlayer was then scrapped off and transferred to conical tubes and centrifuged (3500 g) four times to pellet remnant agar and cellular debris. The supernatant was filtered and concentrated by ultracentrifugation at 100,000 g for 3 h.
DNA isolation and sequencing
DNA was extracted from the prepared high-titer phage stocks using a simple phenol–chloroform protocol. 8 Restriction analysis was performed using different enzymes. Sequencing was performed in a local facility, through the construction of genomic libraries using the Nextera® XT DNA Sample Preparation Kit (Illumina) according to the manufacturer's instructions. Nextera XT Index Kit was used to index individual libraries. Using the Illumina MiSeq platform at the National Institute of Infectious Diseases “Dr. Carlos G. Malbran” (Buenos Aires, Argentina), 143,900 250-bp paired-end raw reads were generated. Adapters and low-quality sequences were trimmed, and Trimmometic v0.36 was used to filter short reads using default settings. 9 After trimming, reads were assembled de novo using Velvet 1.2.10. 10
Bioinformatic analysis and annotation
DNAMaster 5.22.511 was used for sequence analysis and annotation. Subsequently, the information was manually curated. Glimmer 3.0212 and GeneMark 3.25 were used to find the start sites; tRNAs were predicted using Aragorn 1.2.3813 and tRNA-scan 2.0.6,14,15 BLAST, 16 and HHPred 17 were used to assign putative functions.
Transmission electron microscopy
Transmission electron microscopy (TEM) was done in collaboration with the Laboratory of Food Microbiology at ETH Zurich to assess phage morphology. Phosphotungstic acid 2% was used to negatively stain the phage stock for 20 s on a carbon-coated copper grid (Quantifoil), pH = 8.0. The samples were observed at Hitachi HT 7700 scope at 100 kV, equipped with an AMT XR81B Peltier cooled CCD camera (8M pixel).
ImageJ software version 1.51j (NIH, Bethesda, MD) was used to take measurements on 10 individual virions, and then calculate the diameter and length of the capsid and tail.
Results
Lactobacillus phage vB_LcaM_Lbab1 (phage Lbab1) was isolated by enrichment from raw milk obtained from a milking yard from the province of Buenos Aires, Argentina (36° 56′ S, 60° 09′ W), using L. casei BL23 as the isolation host.
Host range and morphology
Plaques formed on L. casei BL23 cells were clear and relatively small with a diameter of 0.39 ± 0.15 mm (Fig. 1A). The host range of phage Lbab1 included L. casei/L. paracasei and L. kefiri strains (Table 1). TEM analysis revealed a myovirus-like morphology (Fig. 1B) with phage particles presenting isometric capsids (103.5 ± 9.6 nm in length; 91.0 ± 3.3 nm in diameter) and tails (161.6 ± 4.5 nm long) with a strikingly complex baseplate structure (Fig. 1C).

Lactobacillus phage Lbab1 has a myovirus-like morphology.
Host Range of Lactobacillus Phage Lbab1
Absence of individual plaques.
Sequencing and genome organization
The Lbab1 genome consists of double-stranded DNA of 140,488 bp in length (486-fold coverage). The genome has a total guanosine cytosine content of 37%, which is significantly lower than that of its host, L. casei BL23 (46.34%). Analysis of the Lbab1 genome revealed 208 potential coding sequences, 202 open reading frames (ORFs), and 6 tRNA genes; 140 ORFs are transcribed rightward and 62 are transcribed leftward. The most used start codon identified was ATG (75.4%), with lower usage observed for GTG (8.4%) and TTG (16.3%). Figure 2 shows a schematic representation of the Lbab1 genome, with a list of predicted genes and their functions given in Supplementary Table S1.
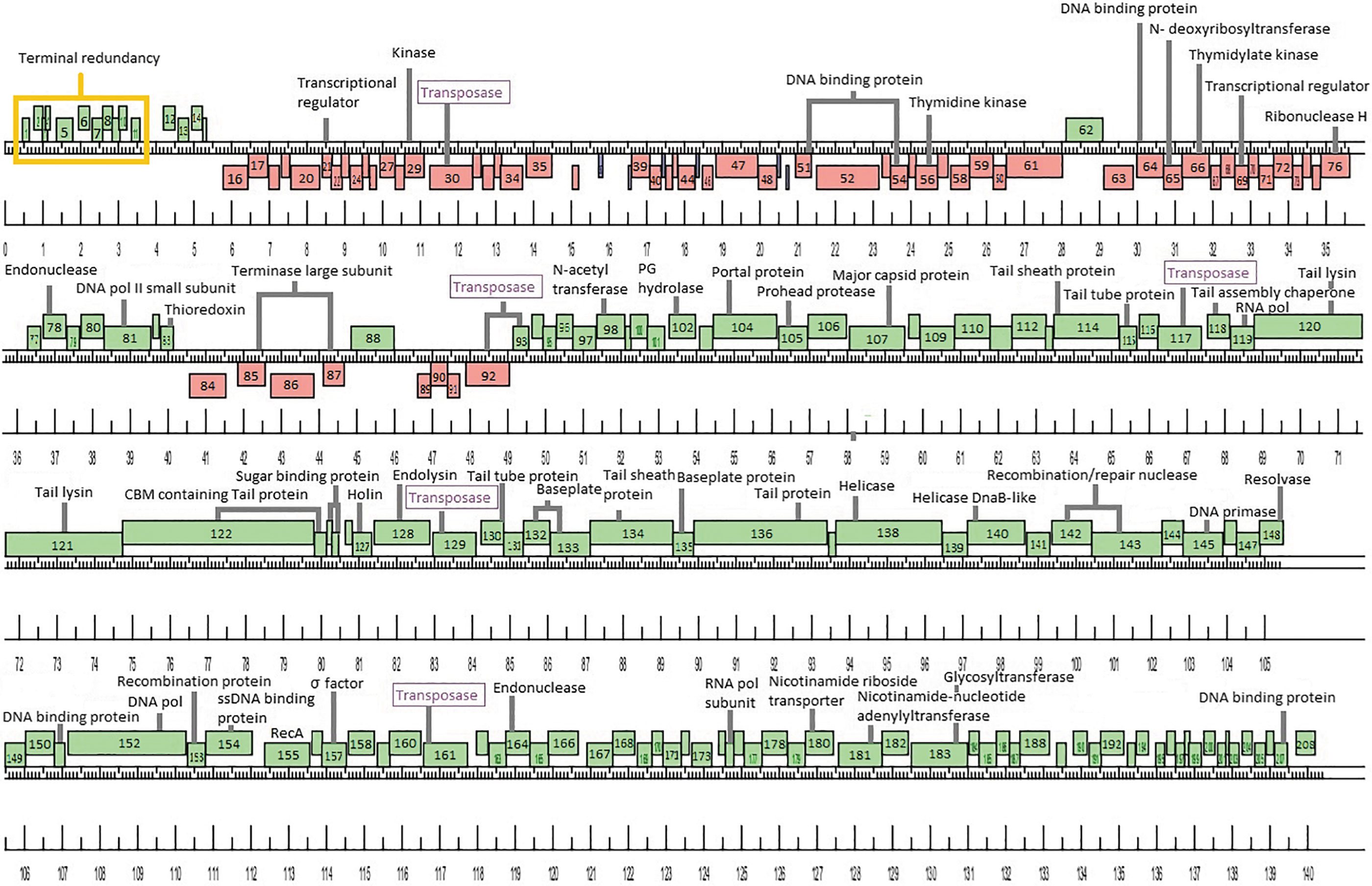
Schematic representation of Lactobacillus phage Lbab1 genome. The viral genome is represented in four tiers, with markers spaced at 100 bp intervals. The predicted genes are shown as boxes either above or below the genome, depending on whether they are rightward (green boxes) or leftward (red boxes) transcribed. tRNAs are shown in blue. Gene numbers are shown within each box.
When analyzing the sequencing data, we noticed that although the average map coverage for the genome was 320 × , there was a region where this increased to 1052 × coverage. This significant increase in sequencing coverage is located between position 80 and 3704 (3624 bp) and is most likely due to the presence of redundant terminal repeats, similar to what was observed for Listeria phage A511. 18 This terminal redundancy includes 11 ORFs, all encoding small gene products without predicted functions.
The complete genome of phage vB_LcaM_Lbab1 is available from GenBank, accession number MT758689.
Comparative genome analysis
The genome of phage Lbab1 exhibits high sequence similarity to Lactobacillus phage Lb338-1 (95.6%), with substantial pairwise span length coverage (82%). Based on its genomic sequence, phage Lbab1 belongs to the Herelleviridae family. A subfamily for Lbab1 and Lb338-1 cannot be assigned. Lactobacillus phage Lb338-1 was isolated in 2009 from a sewage treatment plant located in the south of Ireland using a probiotic cheese strain, L. paracasei NFBC 338, as an isolation host.19,20 Until now, among the fully sequenced genomes of phages infecting L. casei, phage Lb338-1 was the only described myovirus. Since the Lb338-1 genome was extensively analyzed by Alemayehu et al., 19 our analysis of Lbab1 focused on the differences between both phage genomes, as well as predicted additional functions (including previous orphan genes of Lb338-1) to this newly isolated phage.
First, no integrase genes could be identified in the genome of Lbab1, as was previously observed for Lb338-1. In addition, no turbid plaques (a common trait of temperate phages) were observed when infecting L. casei BL23 or any of the other strains tested. Overall, this strongly suggests that Lbab1 maintains a strictly lytic lifestyle. Interestingly, we found six loci with high identity (>80%) to transposases of the IS200/605 family within the Lbab1 genome. This family contains the smallest known DNA transposases (600–1500 bp), which does not feature the characteristic inverted sequences at their ends as found in many prokaryotic and eukaryotic transposons. Instead, these mobile elements carry subterminal sequences, which are able to form hairpin secondary structures at both 5′ and 3′ ends.21,22 Transposases of the IS200/605 family are divided into three major subgroups, based on the presence or absence of two open reading frames: tnpA and tnpB. Although the molecular details of TnpB activity are not known, it does not seem to be required for transposition.23,24 Within the Lbab1 genome, we identified putative transposase coding genes rendering products of ∼380 or 140 amino acids. In one region of the phage genome, we identified both tnpA and tnpB, in opposite orientation (genes 92 and 93). Besides, ORFs 30, 117, 129, and 161 appear to consist of only one transposase. All the transposase coding genes are depicted in Figure 2.
Several structural genes, including those involved in tail formation, were previously identified within the Lb338-1 genome and subsequently confirmed as part of the phage proteome through mass spectrometry analysis of whole viral particles. 19 Unsurprisingly given their high similarity, many homologous genes were also identified within the Lbab1 genome (Supplementary Table S1). We identified a tail/baseplate module spanning genes 114 to 136. Similar to Lb338-1, the lysis cassette containing a putative holin and endolysin (genes 127 and 128) was embedded between the tail structural genes. In phage Lbab1, these two genes are flanked by a complete transposase (gene 129) and a shorter gene 126 encoding a 64 amino acids protein with low identity to gene 49 of Lb-338-1 that also codes for a transposase. This shorter gene product might be nonfunctional. Furthermore, a putative RNA polymerase (gene 119) was found in this region, similar to the Lb-338-1 genome.
The rest of the genes within this region are likely components of the tail and the complex baseplate, as can be observed by TEM. Using HHPred, we also searched for structurally similar domains of these proteins to other known phage proteins. A summary of the results obtained for the putative tail/baseplate proteins is given in Supplementary Table S2. According to this analysis, gp114 and gp115 correspond to the phage sheath and tube proteins, respectively. 25 The predicted structure of gp116 is similar to the LsrR transcriptional regulator with low probability (37, 75%) (PDB 4L5J), suggesting it does not code for a structural virion protein. We believe that gp118 was incorrectly annotated as the tape measure protein in phage Lb338-1. Based on the predicted protein size and BlastP analysis, we strongly believe that gp118 of both phages functions as assembly chaperone. Based on previous Lb338-1 annotations, gene products 120 and 121 are predicted to function as tail lysins. Using HHPred, we identified a short domain with peptidoglycan (PG) hydrolase activity located within the N-terminal region of gp120 and at the C-terminal tip of gp121. 26 For gp122, a fold similar to VgrG1, the needle tip from the bacterial type VI secretion system, 27 was identified within its N-terminus and a carbohydrate binding module (CBM) 28 was found within its C-terminus. The structure of gp123 was predicted to resemble the tail fiber assembly protein of phage Mu and the CBM of a glycoside hydrolase; however, in both cases the probability was ∼50%. Both gp124 and gp125 were predicted to resemble solute binding proteins of a galactobiose ABC transporter 29 (73% and 58% probability, respectively).
Interestingly, when analyzing gene products 114, 115, 130, 132, and 133, we found that for all of them, the most probable structure, usually encompassing almost the whole protein sequence, was highly similar (96–99% probability) to components of the antifeeding prophage (Afp) secretion system of Serratia entomophila. 25 Interestingly, Afp particles are also contractile tail devices with compositions similar to phages, tailocins, and T6SSs. For the rest of the gene products, we only found partial regions with predicted similarity to structural proteins of other Myoviridae phages, as well as to Pyocin R2, the contractile type VI secretion systems, and to various CBMs and proteins with PG hydrolyzing activities.30–32
Conclusions
We have isolated a new Lactobacillus bacteriophage from raw milk. Despite the fact that phage outbreaks can cause significant losses to the dairy industry, only a limited number of Lactobacillus phage genomes have so far been sequenced and annotated. Within public databases, a total of 54 Lactobacillus phages have been sequenced with only 12 known to infect or isolated from L. casei/L. paracasei strains.
From the high-quality images obtained by TEM, a morphological characterization of the phage could be made. Bacteriophage Lbab1 is the second sequenced myovirus infecting L. casei described.
Genome analysis revealed the presence of several transposases of the IS200/605 family identified as possible cause of genetic diversity, promoting rearrangements and genetic exchange observed among other lactic acid bacteria (LAB) phages.
The most similar virus to Lbab1 was phage Lb338-1. Lb338-1 was isolated in 2009 using the probiotic cheese strain L. paracasei NFBC 338 in Ireland, ∼11,000 km from where Lbab1 was isolated. Phage Lb338-1 was isolated in a wastewater treatment plant and Lbab1 was isolated from raw milk. The bacterial host used for isolation of Lbab1, L. casei BL23, has been extensively used as model strain33,34 and proposed as a probiotic due to its anti-inflammatory effects, 35 its ability to prevent colitis in animal models, 36 its ability to modulate host immune responses, 37 and its ability to decrease Listeria dissemination when given orally in mice. 38 The origins of Lb338-1 and Lbab1 are uncertain; however, if these types of phages are present within the mammalian gut, they could potentially reduce the efficacy and beneficial effects of probiotic formulations containing L. casei strains, even if such probiotics are administered daily.
In comparison with other LAB infecting phages, that is, Lactococcus lactis and Streptococcus thermophilus phages, the host recognition machinery of Lactobacillus phages has so far been poorly characterized. Despite their apparent diversity, most of the structures involved in tail and baseplate formation are conserved among phages.39,40 During the past years, an increasing number of phage and phage-like protein structures have been deposited into the Protein Data Bank 41 (rcsb.org), which in combination with structure-based bioinformatics tools such as HHPred are enabling more accurate predictions of structure and function for many (previously unknown) phage components.
The bulky distal tail baseplate observed by TEM and the identification of various CBMs in different structural proteins suggests that phage Lbab1 recognizes cell wall surface polysaccharides as receptors. 40 We have previously shown that L. casei phage J-1 (a syphovirus) uses evolved distal tail CBMs to recognize components of the cell wall polysaccharides during infection42–45 : a characteristic probably extended to other LAB phages.46,47 Nevertheless, additional studies will be necessary to further unravel the functions and structures of all the components involved in host recognition for phage Lbab1 and its relatives. In sum, the expansion of genomic data available for Lactobacillus phages combined with a deeper knowledge about their host recognition machinery could contribute to the development of strategies to avoid infections that cause immeasurable losses in dairy industry.
Footnotes
Authors' Contributions
Lactobacillus phage vB_LcaM_Lbab1 (phage Lbab1) was isolated during the phage course 2016. Owing to time constraints, phage preparations were further purified, subjected to a cesium chloride gradient before TEM analysis and DNA extraction for sequencing. V.G., F.P., M.E.D., L.R.S., E.U., M.D.P., J.P.C., F.Z., and M.A. contributed to concepts/design, data acquisition/analysis/interpretation and article preparation. M.D., P.G., and J.K. contributed to TEM data acquisition/analysis and revising the article. E.S. and D.F.D.P. contributed to phage genome assembly. A.R. contributed to bioinformatics analysis, article preparation, and revising the article. R.R.R. contributed to revising the article. M.P. contributed to concepts/design, data analysis/interpretation, preparation, revising, and approval of the article.
Acknowledgments
Our special thanks to CELFI (Centro Latinoamericano de Formación Interdisciplinaria) (2016–2017), CABBIO (Centro Argentino Brasileño de Biotecnología) (2015), UNU-BIOLAC (United Nations University Biotechnology Programme for Latin America and the Caribbean) (2017), and CABANA (capacity building for bioinformatics in Latin America) (2019) for financial and logistics support and providing travel grants to students and/or invited professors. We are grateful to all the students who participated in the course and isolated new phages. A complete list of the students and affiliations from 2015 to 2019 is provided in ![]() (commentary).
(commentary).
Disclaimer
This article was submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Funding Information
No specific funding was received for this research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.