
Abstract
Background:
There has been a recent resurgence of research on the characterization of Acinetobacter phage for therapeutic use due to the morbidity and mortality associated with treatment failures in cases of multidrug-resistant Acinetobacter baumannii infections.
Materials and Methods:
A bacteriophage isolated from activated sludge that targets A. baumannii ATCC19606 was characterized by electron microscopy, genome sequencing, comparative genomics, and a host range analysis.
Results:
The morphology of Acinetobacter phage DMU1 resembles phages in Siphoviridae. Comparative genomic and phylogenetic analyses reveal that DMU1 is a siphophage and is most closely related to Acinetobacter phage SH-Ab 15497. Out of the strains tested, DMU1 was found to only infect A. baumannii strains ATCC19606 and ATCC17978.
Conclusion:
Acinetobacter phage DMU1 belongs to the Siphoviridae family and is most closely related to Acinetobacter phage SH-Ab 15497. Small-scale host-range analysis of DMU1 indicates a host range that is likely limited to specific A. baumannii strains.
Introduction
Bacteriophages that target Acinetobacter spp. have been reported in the literature for many years and were initially used for the genetic manipulation of Acinetobacter using transduction and for classification of Acinetobacter strains via phage typing.1–3
Research on bacteriophages that infect and kill Acinetobacter baumannii has intensified over the past several years due to well-founded concerns regarding antibiotic resistance and the need for alternative treatment strategies. To date (October 2020), ∼110 complete genomes of phage that target A. baumannii are publicly available, divided among only four families. Most of these phages belong to Myoviridae and Autographiviridae, with 49 and 42 phages, respectively. The remainder of these Acinetobacter phages are split between two families with 16 phages in Siphoviridae and 1 in Ackermannviridae. These phages comprise only a small fraction of characterized bacteriophages that target medically relevant bacteria. The urgent need for new treatment options to combat multidrug-resistant A. baumannii and the paltry number of characterized phages that kill these pathogens underscore the need to discover and characterize new Acinetobacter phages.
Herein, we report the isolation and characterization of Acinetobacter phage DMU1, a lytic bacteriophage that belongs to the Siphoviridae family.
Materials and Methods
Bacterial culture
Cultures were initiated from bacterial glycerol stocks kept at −80°C. Strains were streaked on to Lysogeny broth (LB) agar (Miller) and incubated at 37°C for ∼16 h. To obtain log-phase liquid cultures, single colonies were resuspended in LB, and incubated at 37°C, 180 rpm for ∼16 h followed by dilution 1/100 in LB and further incubation at 37°C, 180 rpm for 2 h. Bacterial strains A. baumannii ATCC19606, A. baumannii ATCC17978, A. baumannii AYE, A. baylyi ADP1, and Moraxella catarrhalis ATCC25238 were obtained from the American Type Culture Collection. A. baumannii RUH134 and A. nosocomialis M2 were a gift from Robert S. Munson, Jr.
Phage isolation
Activated sludge was obtained from the Des Moines Metropolitan Wastewater Reclamation Authority for use in the isolation of bacteriophages as previously described, with modifications. 4 The activated sludge was clarified via centrifugation at 8000 g for 10 min and then filtered sequentially with 0.45 and 0.22 μm sterile aPES bottle-top vacuum filters (Thermo Fisher, Waltham, MA). For enrichment, 10 mL of clarified sample was mixed with 10 μL of an overnight culture of A. baumannii ATCC19606 and 40 mL of LB (Becton, Dickinson, and Company, Sparks, MD). This enrichment culture was incubated at 37°C, 180 rpm overnight. A 10 mL aliquot of this enrichment culture was clarified and filtered as described above using aPES syringe filters. The agar overlay method was used to culture phages for isolation of individual plaques. 5 A 5 mL aliquot of molten top agar (0.6% agar w/v) was cooled to ∼45°C, mixed with 100 μL of the clarified filtered lysate and 10 μL of an overnight culture of A. baumannii ATCC19606, and was poured onto a room temperature LB agar plate. After overnight incubation at 37°C, individual plaques were isolated and restreaked as previously described. 6 Briefly, the center of a plaque was punched out with a transfer pipette and the agar plug was resuspended in saline magnesium (SM) buffer comprised 100 mM NaCl, 25 mM Tris-HCl pH 7.5, 8 mM MgSO4, and 0.01% (w/v) gelatin. Then, 10 μL of the isolated plaque mixture was spotted onto an agar overlay containing A. baumannii ATCC19606 and streaked using a sterile filter paper strip. These plates were incubated at 37°C overnight. This process of picking and streaking phages was repeated a total of three times.
High-titer lysate-isolated phages were obtained from multiple agar overlay plates as previously described with modifications. 7 Briefly, top agar overlays were prepared as described above to obtain five plates with near-confluent plaquing. The overlay was broken apart with a plate spreader. A slurry created with SM buffer and the disturbed agar overlays was collected into a 50 mL conical tube and centrifuged at 8000 g for 10 min. The supernatant was decanted and clarified using syringe filters, as described above, and stored at 4°C.
Analysis of phage morphology by transmission electron microscopy
Phages were prepared for transmission electron microscopy (TEM) by sedimentation as previously described with modifications. 8 Briefly, 1 mL of high-titer phage lysate was sedimented in a microcentrifuge for 90 min at 21,000 g. The supernatant was discarded, and the pellet was washed with 1 mL of 0.1 M ammonium acetate solution, pH 7, and sedimented again for 90 min at 21,000 g. This step was repeated one additional time, and ∼10 μL of ammonium acetate was left in the tube with the final sediment. Carbon/formvar grids were treated with 0.1% bacitracin (Fisher Scientific) in H2O for 2 min to make the grids more hydrophilic. A piece of filter paper was applied to an edge of each grid to remove excess liquid; grids were placed on a drop of phage suspension (diluted 1:1 with H2O) and allowed to adsorb for 2 min. Excess liquid was removed, as described above. Phages were negatively stained by floating the grids on a drop of 2% uranyl acetate (Courtesy of Central Microscopy Research Facility at the University of Iowa) for 1 min. Excess liquid was removed again, and grids were air-dried for 10 min. The phages were imaged using a JEOL JEM-1230 TEM at the Central Microscopy Research Facility at the University of Iowa. Approximately 60 phage particles were examined using ImageJ 9 to measure the length and width of phage tails and the diameter of phage capsids.
DNA isolation
Genomic DNA was isolated by using a phenol/chloroform-based method. Briefly, phage lysates were mixed with 10% PEG-800 and 1 M NaCl at a 2:1 ratio, and were incubated overnight at 4°C or placed on ice for 60 min. After incubation, samples were centrifuged at 10,000 g for 30 min. Pellets were resuspended in 500 μL 5 mM MgSO4 by pipetting. DNase1 (50 U) and RNaseA (25 μg) were added, and samples were incubated at 37°C for 1 h. Proteinase K (25 μg), 25 μL of 10% sodium dodecyl sulfate, and 20 μL of 0.5 M ethylenediaminetetraacetic acid, pH 8.0, were added, and samples were incubated at 60°C for 1 h. After cooling to room temperature, phenol:chloroform:isoamyl alcohol pH 8.0 was added at a 1:1 ratio. Samples were gently mixed by inversion and centrifuged at 3000 g for 5 min at room temperature. The phenol–chloroform extraction was repeated until the aqueous layer was no longer cloudy. Chloroform was added to the solution, gently mixed by inversion, and the tubes were centrifuged as described above. DNA was precipitated using sodium acetate and ethanol as previously described. 10 Precipitated DNA was resuspended in 50 μL of IDTE (Integrated DNA Technologies, Coralville, IA). Quality control of the DNA samples was performed using spectrophotometry (NanoDrop; Thermo Fisher) and fluorometry (Qubit; Thermo Fisher).
DNA library prep and MinION sequencing
A MinION sequencing library was prepared using the purified native ligation sequencing kit (SQK-LSK109; Oxford Nanopore), native barcoding expansion 1–12 kit (EXP-NBD104; Oxford Nanopore, United Kingdom), and the NEBNext Companion Module for Oxford Nanopore Technologies Ligation Sequencing (New England BioLabs, Ipswich, MA) as per the manufacturer's instructions. DNA was purified using CleanNGS SPRI beads (Bulldog Bio, Portsmouth, NH). DNA was quantified using a Qubit 2.0 fluorometer in conjunction with the Qubit HS DNA Assay Kit (Thermo Fisher) as per the manufacturer's instructions. The prepared library was sequenced for ∼2 h using an R10 flow cell on the MinION platform (v19.12.2; Oxford Nanopore).
Genome assembly and annotation
A portion of the gDNA sample was submitted to the University of Minnesota Genomics Center for library generation and sequencing using an Illumina MiSeq. Libraries were generated using the Nextera XT kit as per the manufacturer's instructions and were sequenced with the MiSeq system using the MiSeq Reagent Kit v2 Nano with a 2 × 150 bp read length.
Bioinformatics
Raw data from the MinION device was base called by using Guppy (v4.0.14; Oxford Nanopore) with default settings. Reads smaller than 1 kb were excluded from further analysis using Filtlong (v0.2.0, github.com/rrwick/Filtlong). Demultiplexing and barcode and adapter trimming were performed using Guppy. An initial assembly of the MinION data was performed using Canu 11 (v2.0) with standard settings and Medaka v1.0.3 (github.com/nanoporetech/medaka).
Raw paired-end Illumina MiSeq reads were processed using BBtools Dedupe (v38.86, sourceforge.net/projects/bbmap/) to remove duplicate reads. Adapter trimming was performed using Trimmomatic 12 (v0.39) using default settings for paired-end reads.
Unicycler 13 (v0.4.8) was used with standard settings to form a hybrid assembly of both the short 2 × 150 bp Illumina reads and the MinION long reads. Unicycler has several dependencies that were used in this analysis, including Pilon 14 (v1.23), Samtools 15 (v1.10), Bowtie216 (v2.3.5.1), Racon 17 (v1.4.11), and SPAdes 18 (v3.14).
Tools made available through the Center for Phage Technology (CPT) at Texas A&M University, namely Galaxy and WebApollo instances, were used for genome annotation. 19 Structural annotation was performed using the CPT PAP Structural Workflow (v2020.01), which uses Glimmer320 (v3.02b) and MetaGeneAnnotator 21 (v1.0) to call open reading frames, Aragorn 22 (v1.2.36) for tRNA prediction, and TransTermHP 23 (v2.0.9) for transcriptional terminator prediction. These data were then used for functional annotation using the CPT PAP Functional Workflow (v2020.04). This annotation pipeline performs nucleotide/amino acid similarity searches (blastn, blastp, InterProScan) to identify related genes, proteins, or protein families and uses algorithms to predict transmembrane domains (TMHMM), 24 and lipidation signals (LipoP, 25 v1.0). This information, along with the structural annotation, was used to predict and assign functional identities to genes and proteins. The genome was visualized using GCView.26,27 The complete genome sequence of Acinetobacter phage DMU1 reported here was deposited in GenBank under accession number MT992243. Basecalled reads can be found in the Sequenced Read Archive under accession numbers SRR12622056 and SRR12222057.
Comparative genomics and phylogenetics
Blastn, progressiveMauve algorithm of Mauve 28 (v2.4), and FastANI 29 (v1.32) were used for comparative genomics. Phylogenetic analysis was performed on the isolated phage genome and a select number of Acinetobacter phages (Supplementary Table S1), as described previously. 30 Briefly, phage protein sequences were clustered into orthologous groups using OrthoMCL 31 (v2.0.9). A binary matrix was created based on representation in a cluster and used for Jaccard similarity and distance calculation. These data were visualized with Splitstree 32 (v4.16.1) using the Neighbor-net method. 33
Host range
Host range determination was performed as previously described 34 by spotting diluted phage stocks on agar overlays containing A. baumannii ATCC19606, A. baumannii ATCC17978, A. baumannii AYE, A. baumannii RUH134, A. nosocomialis M2, A. baylyi, or M. catarrhalis ATCC25238. Plates were incubated overnight at 37°C, and bacterial lawns were examined for a zone of clearing where the phage isolate was spotted. A total of three independent replicates were performed, all on different days.
Results
Bacteriophage DMU1 was isolated from activated sludge obtained from the Des Moines Metropolitan Wastewater Reclamation Authority using A. baumannii ATCC19606 as the host. In agar overlays of A. baumannii ATCC19606 and DMU1 incubated ∼16 h, DMU1 produces clear plaques ≤2 mm in diameter with defined edges.
Observation by TEM revealed that DMU1 phage virions possess icosahedral heads and long transversely striated noncontractile tails, which appear to have tail spikes or fibers at the terminal end (Fig. 1A). The tails of a significant number of observed phage particles appear slightly curved, indicating a flexible tail. The phage capsid of DMU1 measured 63.36 ± 1.9 nm, and the tail was 9.61 ± 0.7 nm wide and 114 ± 5.35 nm long. The morphological features of the phage isolates discussed above are consistent with belonging to the Siphoviridae family.
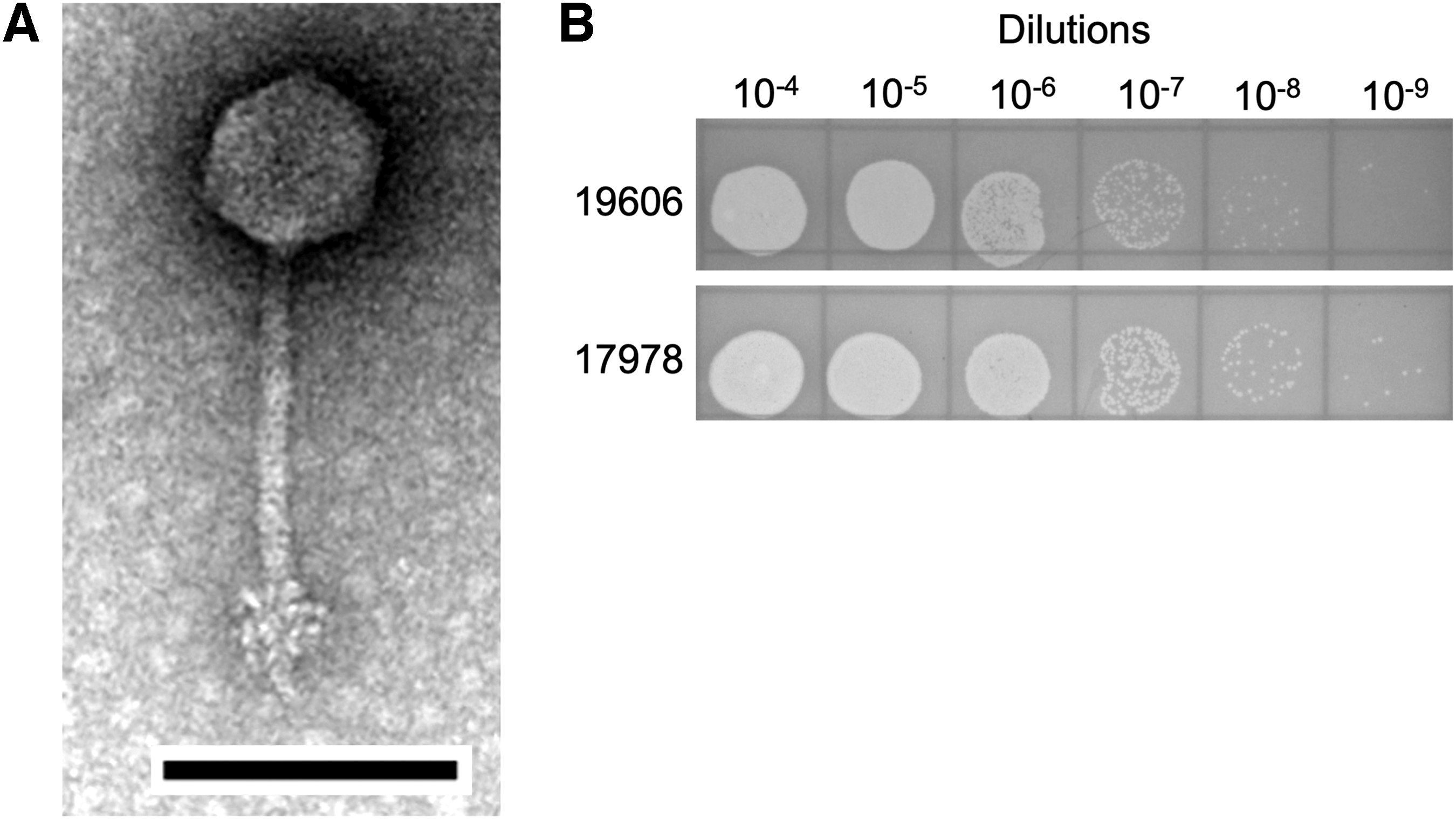
In our host range analysis, DMU1 was only observed to produce plaques in agar overlays of A. baumannii ATCC19606 and A. baumannii ATCC17978 (Fig. 1B). No observable plaques were seen on soft agar overlays containing A. baumannii AYE, A. baumannii RUH134, A. nosocomialis strain M2, A. baylyi ADP1, and M. catarrhalis ATCC25238.
DNA sequencing of DMU1's genome using both Oxford Nanopore and Illumina platforms provided 5504x coverage of a circular genome of 43,482 bp with a 47.86% G + C content. The MinION nanopore reads and Illumina MiSeq 2 × 150 bp reads provided an average coverage of 579x and 4925x, respectively. DMU1 is predicted to have a coding density of 96%, with 60 genes that are predicted to encode for 59 proteins and one arginine-tRNA. Also, only a single transcriptional terminator was predicted (Fig. 2). Functional annotation of the genome resulted in predictions for 20 proteins encoded on the Acinetobacter phage DMU1 genome. Proteins with predicted functions in DNA packaging and host cell lysis, in replication and nucleotide metabolism, and as structural phage elements were identified. In total, our functional annotation was unable to predict functions for 36 of the 59 (61%) proteins encoded within the DMU1 genome. No lysogeny-related genes such as integrases were annotated.
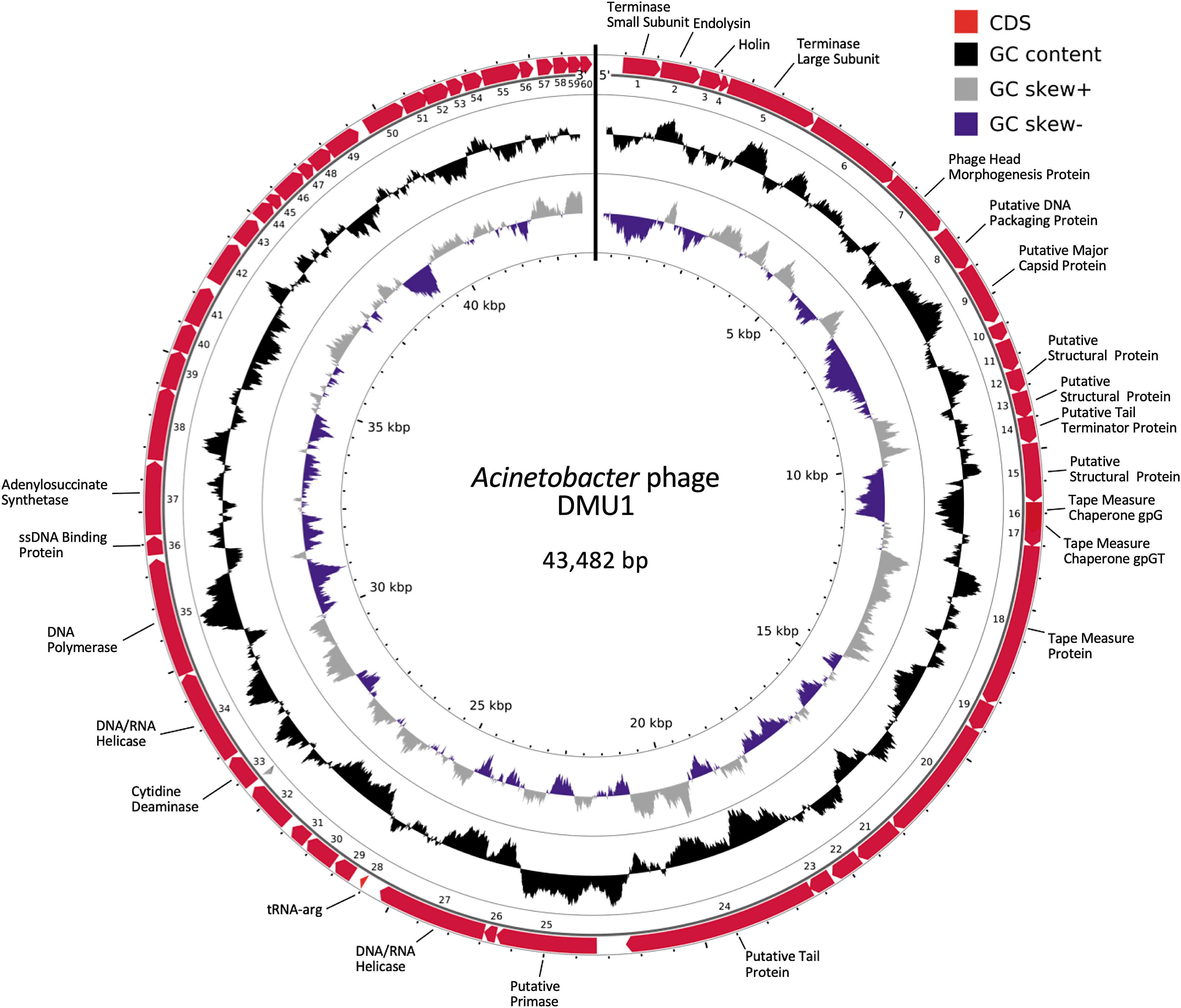
Acinetobacter phage DMU1 genome map. The outer ring illustrates the coding sequences (CDS) of the genome (cardinal). Genes are labeled with predicted functions for the proteins they encode. The inner rings show G + C content (black), and GC skew positive (gray) and negative (purple).
Blastn of Acinetobacter phage DMU1 genome sequence revealed only one significant hit, which was the genome of Acinetobacter phage SH-Ab 15497. 35 Global alignments between the genomes of DMU1 and SH-Ab 15497 were performed using a global blast alignment (National Center for Biotechnology Information) and Mauve. Blast global alignment results show a 92% identity (40,243/43,784 bp) with 666 gaps. The average nucleotide identity between DMU1 and SH-Ab 15497 was estimated by FastANI to be 95%, which indicates that these phages belong to the same species. The Mauve alignment between these two genomes reflects the global alignment results showing a high degree of genome synteny as both genomes are contained within a locally colinear block (Fig. 3). A network phylogeny was generated based upon the orthologous protein groups identified by OrthoMCL using the proteins encoded within 77 selected Acinetobacter phage genomes. This analysis places DMU1 near other Acinetobacter siphophages (Fig. 4).
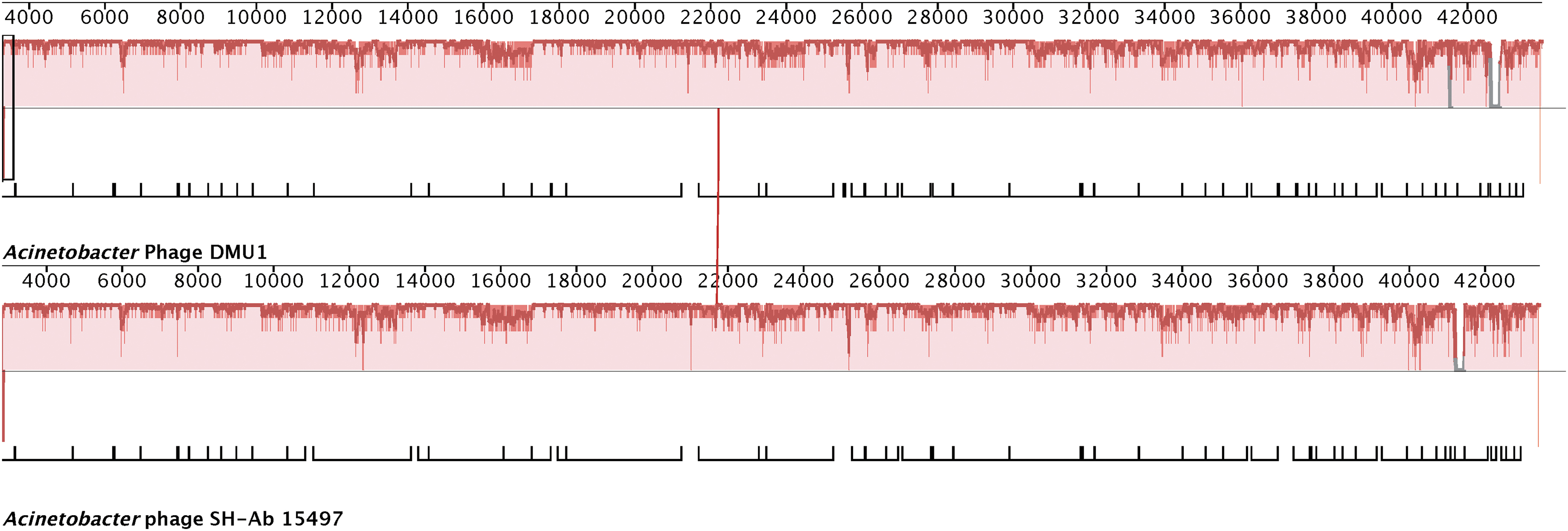
progressiveMauve alignment of Acinetobacter phages DMU1 and SH-Ab 15497 genomes. The X axis represents genome position and Y axis (red bars) represents nucleotide sequence similarity. Gaps indicate insertions in phage's genome. Annotated genes for each genome are depicted as white blocks below the similarity plots.
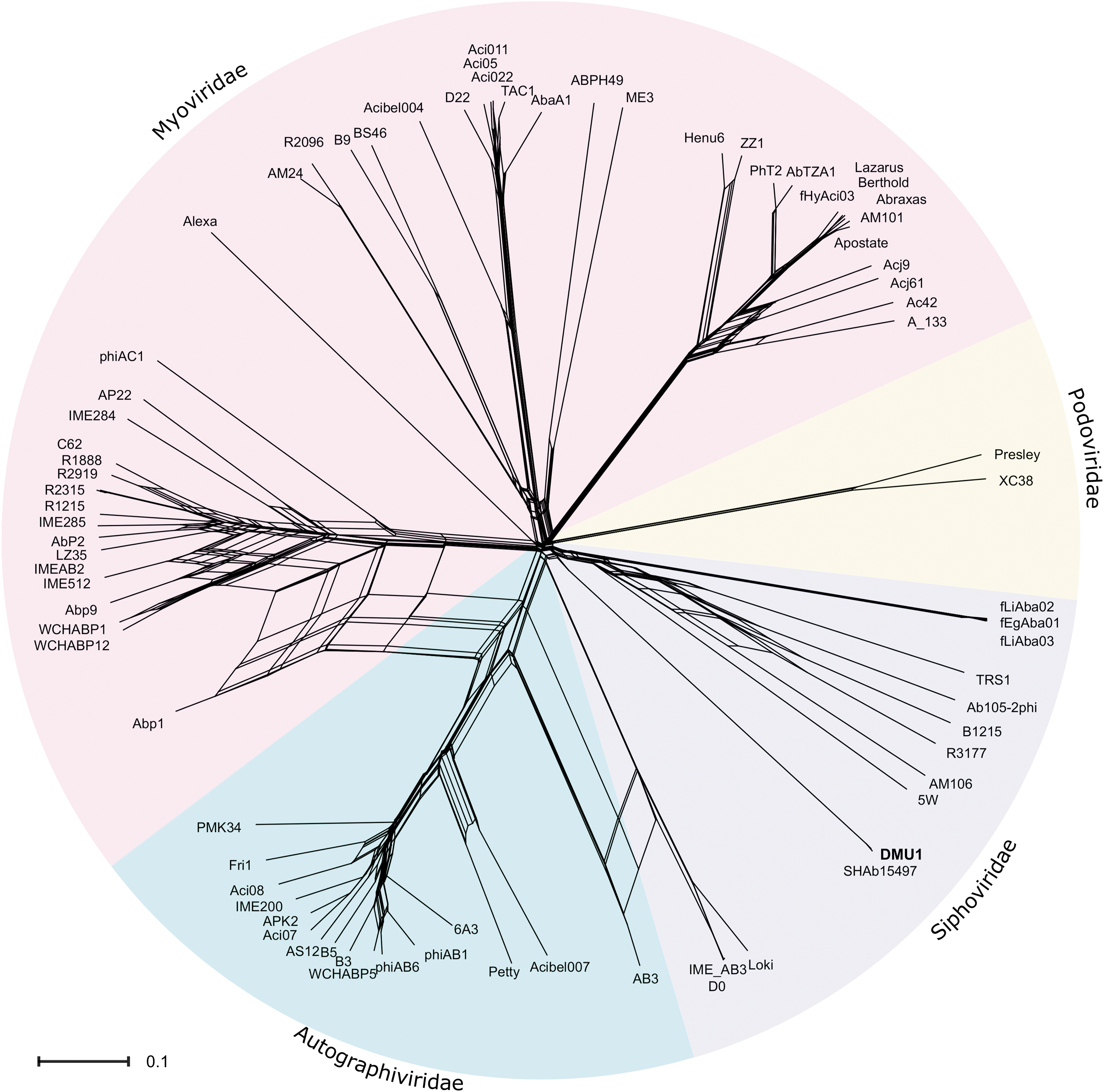
Net neighbor split graph based on Jaccard distances between the proteins encoded by 76 Acinetobacter phage genomes. Node labels represent individual phage. Labels and the corresponding GenBank/RefSeq accession numbers are located in Supplementary Table S1. The scale bar represents the number of protein differences (presence or absence) per protein site.
Discussion
A phage, called DMU1, was isolated against A. baumannii ATCC19606 from activated sludge. Observation of DMU1 virions by TEM revealed symmetrical icosahedral capsids with long transversely striated flexible tails that appear to terminate with tail spikes and/or fibers. These observations, along with the measured dimensions of DMU1, are consistent with phages in the family Siphoviridae. Host range analysis, while limited in the number of A. baumannii strains, points to DMU1 exhibiting a narrow host range. Additional phenotypic analyses, such as one-step growth and kill curves, have been attempted. When our preparations of DMU1 are added to log-phase A. baumannii ATCC19606 cultures, we see an almost immediate and marked decrease in A600 that is sustained for several hours without an increase in plaque forming units (PFU) (data not shown). Changing our methods to generate DMU1 lysates or further purification of DMU1, to avoid this overt toxicity, has not been fruitful.
The genome length of Acinetobacter phage DMU1 was determined to be 43,482 bp, which is consistent with Acinetobacter siphophages reported in a comparative genomics study by Turner et al., with an average length of 43,000 bp. 30
The predicted protein functions encoded by annotated genes on the Acinetobacter phage DMU1 genome can be loosely grouped into three main categories, which are lysis-associated proteins, structural proteins, and DNA replication proteins. We note that our annotation did not identify genes encoding i-, o-, or u-spanins and/or disruptins.
The genome of DMU1 shares extensive identity with a previously characterized Acinetobacter siphophage, SH-Ab 15497, isolated in Shanghai, China. 35 Acinetobacter phage SH-Ab 15497 was isolated from hospital sewage against an unnamed A. baumannii clinical isolate and was reported to infect 71/234 isolates. In a much smaller panel of hosts, DMU1 was able to infect 2/4 A. baumannii isolates and 0/3 of other gram-negative bacteria. The genome lengths and G + C content of DMU1 and SH-Ab 15497 are nearly identical (43,482 bp, 47.83% G + C and 43,420 bp, 47.86% G + C, respectively).
Phylogenetic analysis based on orthologous groups of proteins indicates that DMU1 is related to other Acinetobacter siphophages and thus should be classified within Siphoviridae. Unsurprisingly, DMU1 and SH-Ab 15497 are clustered together. However, both form a distinct cluster apart from other Acinetobacter siphophages and thus likely represent a new genus.
In summary, Acinetobacter phage DMU1, a phage belonging to the Siphoviridae family, was isolated from activated sludge and characterized. This work adds to the growing knowledge of bacteriophage research and their characteristics. More phages targeting A. baumannii should be isolated and characterized to better understand the mechanisms by which bacteriophages target and kill their host, to build better phage collections, and ultimately for the potential use in phage cocktails for therapeutics of A. baumannii infections.
Footnotes
Authors' Contributions
M.D.C., B.M.P., and F.E.K. designed the experiments. M.D.C., B.M.P., F.E.K., and D.N. performed the experiments, analyzed data, and contributed to this article's writing. All authors have reviewed and approved the article before submission and agree to be accountable for all aspects of the work. The funders had no role in the study design, data collection, or in the decision to submit this work for publication.
Acknowledgments
Special thanks go to Tim Runde, Larry Hare, and the Des Moines Metropolitan Wastewater Reclamation Authority for providing the activated sludge for this project; Tom Moninger at the University of Iowa Central Microscopy Research facility for technical assistance with TEM; and Aaron Becker along with the staff at the University of Minnesota Genomics Center who performed the Illumina library preparation, quality control, and Mi-Seq sequencing.
Disclaimer
This article is an original work that has been submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by start-up funds from the Iowa Osteopathic Education and Research Fund and Iowa Academy of Science grant #18-07 awarded to M.D.C.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.