
Abstract
Introduction:
Antibiotic resistance and virulence are common among bacterial populations, posing a global clinical challenge. The bacterial species Acinetobacter pittii, an infectious agent in clinical environments, has shown increasing rates of antibiotic resistance. Viruses that integrate as prophages into A. pittii could be a potential cause of this pathogenicity, as they often contain antibiotic resistance or virulence factor gene sequences.
Methods:
In this study, we analyzed 25 A. pittii strains for potential prophages. Using virulence factor databases, we identified many common and virulent prophages in A. pittii.
Results:
The analysis also included a specific catalogue of the virulence factors and antibiotic resistance genes contributed by A. pittii prophages. Finally, our results illustrate multiple similarities between A. pittii and its bacterial relatives with regard to prophage integration sites and prevalence.
Discussion:
These findings provide a broader insight into prophage behavior that can be applied to future studies on similar species in the Acinetobacter calcoaceticus-baumannii complex.
Introduction
A
One mechanism of A. pittii pathogenicity could be through prophages. Prophages are formed by bacteriophages when they integrate their entire genome into bacterial genetic sequences during a lysogenic life cycle, and these prophages propagate by replicating with bacterial chromosomes or plasmids during cell division. 9 Through lysogeny, prophages can spread throughout a bacterial population without killing its host.
This allows prophage propagation to not only benefit the phage but also confer advantages on the host bacterium, such as increased genetic diversity 9 and the prevention of additional viral infection. 10 Moreover, prophages often contain virulence factors (proteins that enhance the pathogenicity of their host) and, thus, increase the survival fitness of both phage and bacteria. 9
Prophages have also been associated with virulence and antibiotic resistance in the Acinetobacter genus.11,12 In A. baumannii, prophages have been found in bacterial chromosomes and plasmids that encode a variety of virulence factors ranging from efflux pumps that remove toxins to antibiotic-inactivating enzymes. 11 However, although related species such as A. baumannii have been surveyed for integrated viral sequences, such prophage analysis has yet to be applied to A. pittii.
Given the clinical infections resulting from A. pittii, it is imperative that the mechanisms and origins of its pathogenicity be better understood. As a result, by identifying the specific prophages and virulence factors that are most common in A. pittii, this study could provide new directions for A. pittii research and pinpoint existing or potential future causes of its virulence.
In our study, we characterized the distribution, lengths, and relative genome positions of prophages found in A. pittii. We found that there is a clear but uneven distribution of prophages in A. pittii strains, and that the prophages exhibit differences with respect to viral families. To further understand their influence on bacterial hosts, we searched for and analyzed virulence factors and antibiotic resistance genes within these prophages. Our results indicate that virulence factors, in general, compared with antibiotic resistance genes, are present in greater quantities and varieties. In addition, we present evidence of virulence factors potentially influencing genomic diversity and rearrangements.
Materials and Methods
Genome collection
Our study used only complete A. pittii genomes (24) and chromosomes (1), which were obtained from NCBI GenBank (last accessed January 2021). 13 Each genome or chromosome belonged to a different strain of the bacteria. Genomes consisted of the fully sequenced chromosome for that specific strain as well as all associated plasmids. As a result, there were in total 25 chromosomes and 64 plasmids across the 25 A. pittii strains.
Prophage identification
Using the GenBank accession number for each chromosome or plasmid, as well as the PHAge Search Tool-Enhanced Release (PHASTER) web server, 14 we identified potential prophages within each genetic sequence. To ensure high confidence in the identity of sequences as potential prophages, those that were less than 10 kb in length or did not contain structural genes and/or integrase were discarded, as per Costa et al. 11
The selected prophages were then automatically classified as Intact, Questionable, or Incomplete by the PHASTER software based on the amount of phage genes in the prophage sequence. 14 This automatic classification depends on factors such as amount of phage coding regions and sequence length. 14 PHASTER also classified each non-discarded prophage as belonging to the Siphoviridae, Myoviridae, or Podoviridae viral families. In total, 94 prophages were identified for the A. pittii strains, of which 34 were labeled Intact by PHASTER. Due to their completeness, only these 34 intact prophage sequences were used for the analysis steps described next.
Statistical and graphical analysis
Length data were retrieved from PHASTER for each of the 34 intact prophages. Statistical analysis of average prophage length within each viral family was then performed in Microsoft Excel (2019) by using a one-tailed t-test, assuming unequal variances. The significance level was taken to be p < 0.05.
Graphical analysis on the spatial distribution of intact prophages within bacterial chromosomes was performed by using data from PHASTER and the R package ggplot2. 15 Specifically, the geom_density() function of ggplot2 was employed to create a density plot of the intact prophages along bacterial chromosome sequences.
Virulence factor identification
Chromosomes and plasmids with at least one intact prophage were selected for genome annotation by using the myRAST annotation software and standard parameters. 16 Thus, all protein encoding genes within the 34 intact prophages were identified and run in a BLAST search against the Virulence Factor DataBase (VFDB) with default parameters and the protein sequences from VFDB's full dataset. 17 An Expect value of less than 1 × 10−20 was used as a cutoff, and virulence factors were thus identified among the intact prophages.
Synteny comparison
Two of the most common virulence factors, as identified by VFDB, were further analyzed. These were anthranilate phosphoribosyltransferase and zinc binding alcohol dehydrogenase, each present in three intact prophages. Synteny maps for each set of prophages were generated by using Mauve 18 to compare the virulence factors shared within the set.
Resistance gene identification
The sequence for each of the intact prophages was inputted into the Resistance Gene Identifier of the Comprehensive Antibiotic Resistance Database (CARD). 19 By comparison with CARD, potential antibiotic resistance genes were identified within the prophages. Each resistance gene was labeled by CARD as Perfect, Strict, or Loose depending on the degree of confidence for the presence of that gene. “Perfect” sequences exactly matched an existing antibiotic resistance gene sequence in CARD, whereas Strict and Loose results were less exact matches. 19
Phylogenetic comparison
We constructed a phylogenetic tree comparing the Acinetobacter phage YMC/09/02/B1251_ABA_BP in A. pittii and A. baumannii strains. We retrieved the GenBank sequences for intact A. pittii prophages identified in our study, as well as the GenBank sequences for intact A. baumannii prophages. The 20 intact prophages were aligned by using MAFFT version 7 and default parameters. 20 The phylogenetic tree was then generated by using MEGA, 21 with bootstrap replications set to 100.
Results
Prophage presence
The complete genomes of 25 A. pittii strains available on NCBI GenBank in January 2021 were retrieved. These included 25 bacterial chromosomes (one per strain) and 64 plasmids (Supplementary Table S1). Using PHASTER, 119 potential prophages were identified within 17 of the 25 A. pittii strains. Of the 119 potential prophages, 25 were discarded due to low confidence in their prophage identity, either because of short sequence length or due to lack of common phage genes such as integrase. Out of the remaining 94 prophages used for the analysis, we had (in decreasing degree of confidence in prophage identity) 34 intact prophages, 29 questionable prophages, and 31 incomplete prophages.
Table 1 and Supplementary Table S2 shows the distribution and prevalance of the 34 intact prophages across the 17 A. pittii strains, which contained these high-confidence sequences. Intact prophages were distributed unevenly across the strains, with the strains ST220, HUMV-6483, and WCHAP005069 containing the most intact prophages (four each). The prevalence of these prophages in the different A. pitti strains is shown in Supplementary Figure S3.
Intact Prophage Distribution Across Acinetobacter pittii Strains
In terms of genetic element type, 2 out of 64 bacterial plasmids contained intact prophages, whereas 17 out of 25 bacterial chromosomes contained intact prophages. For these 19 sequences (2 plasmids and 17 chromosomes) that contained intact prophages, there averaged 1.00 prophages per plasmid and 1.88 prophages per chromosome.
Prophage distribution and characteristics
All 94 prophages (intact, questionable, and incomplete) were predicted, by PHASTER and GenBank comparison, to belong to the viral order Caudovirales and the viral families Myoviridae, Siphoviridae, or Podoviridae. Of the three viral families, Siphoviridae accounted for most of the total prophages (65%, 61 out of 94), but Myoviridae accounted for the majority of intact prophages (56%, 19 out of 34). Podoviridae was present in the smallest amount, accounting for only 5 out of 94 total prophages and none of the intact prophages.
To analyze the effects of A. pittii prophage genome breakdown after integration, we compared the average lengths of intact Myoviridae and Siphoviridae prophages. Siphoviridae sequences averaged 49.3 kb in length, whereas Myoviridae sequences averaged 37.1 kb. Using a one-tailed t-test assuming unequal variances, the lengths of the Siphoviridae were found to be significantly greater than those of Myoviridae.
Using the geom_density function of R package ggplot2, 15 a density plot for the intact prophages present in A. pittii chromosomes was generated (Fig. 1). Prophages present in plasmids were omitted due to the genetic differences and shortened lengths of these mobile genetic elements. The density plot demonstrated that there was significant prophage density around 1.4 and 3.2 Mbp, with the 3.2 Mbp peak having the higher prophage density. It can be seen in Figure 1 that there were many prophage sequences starting and ending at these two peaks. Thus, the preferential insertion of prophages within these two regions could aid in the identification of novel prophages in other bacterial strains.

Spatial distribution of prophages in Acinetobacter pittii genomes. A density plot was generated in R for all intact prophages within A. pittii chromosomes (n = 32). The x-axis indicated positions along the A. pittii genome in base pairs, whereas the y-axis indicated the probability of containing prophages per base pair unit. The area under the curve for an x-axis interval, thus, provided the prophage density for that particular genome region. The resulting plot showed increased prophage density at two peaks along the bacterial chromosome: 1.4 and 3.2 Mbp. The red line denotes the density in terms of prophage start locations, whereas the green line denotes the density in terms of prophage end locations. Both show similar peaks.
Hereafter, analysis is performed only with the 34 intact prophages, which are referred to as simply “prophages.”
Prophages contain antibiotic resistance genes
Using the Resistance Gene Identifier of the CARD, 19 37 antibiotic resistance genes were identified within 19 of the 34 intact prophages (Table 2). Resistance genes were classified as Loose, Strict, or Perfect within CARD in increasing degrees of similarity between the prophage gene and the CARD sequence. 19 The majority (73.0%), were classified as Loose, although the remaining 10 resistance genes did fulfill the Strict or Perfect criteria. Interestingly, nine out of these 10 genes were found in the Burkholderia phage phiE12-2 present in the C54 strain's plasmid, which was also the prophage that contained the most virulence factors.
Presence of Antibiotic Resistance Genes Within Acinetobacter pittii Prophages, as Classified by the Comprehensive Antibiotic Resistance Database
AAC, aminoglycoside acetyltransferase; ABC, ATP-binding cassette; ARO, Antibiotic Resistance Ontology; CAT, chloramphenicol acetyltransferase; CRP, cyclic AMP receptor protein; IMP, active on imipenem; LRA, β-lactam resistance Alaskan; MFS, major facilitator superfamily; MPH, Macrolide phosphotransferase; RND, resistance-nodulation-cell division; SPG, Sphingomonas.
Of the 37 resistance genes (Table 2), efflux pumps constituted the largest category of resistance genes (48.6%). These proteins remove toxic substances, including antibiotics, from the cell interior. 22 Notably, adeL genes were present in the highest quantity (13.5% of total resistance genes). adeL is significant, because it regulates the expression of the AdeFGH Resistance Nodulation Division efflux pump system, which can confer multidrug resistance that includes resistance to tetracycline, trimethoprim, and chloramphenicol.23,24
Virulence factors within the prophages
Using VFDB, 47 protein-encoding genes were identified in the prophages as encoding putative virulence factors (Table 3). The most commonly occurring virulence factors were invasion plasmid antigens (7 out of 47 virulence factors) and IS6 family transposases (5 out of 47). However, some virulence factors—such as the IS6 family transposases—were present numerous times in only one or a few of the 34 prophages. As a result, the virulence factors present in the most prophages were invasion plasmid antigens (4 out of 34 prophages), anthranilate phosphoribosyltransferases (3 out of 34), and zinc binding alcohol dehydrogenases (3 out of 34).
Virulence Factor Distribution in 34 Acinetobacter pittii Prophages, as Classified by the Virulence Factor Database
Identified virulence factors were all part of Virulence Factor DataBase.
We categorized the virulence factors by their broader functions, based on a literature review (Table 4 and Supplementary Fig. S4). Based on functional categories, proteins involved in cellular metabolism and biosynthesis made up the largest fraction (12 out of 47 virulence factors, 25.5%). These proteins, thus, regulate key nutritional and energetic requirements in the cell that would help bacteria respond to stressful environments. The individual prophage (not species) that contained the most virulence factors was the Burkholderia phage phiE12-2 in the C54 strain's plasmid (Table 3). It contained nine out of 47 total virulence factors (19.1%) within its genetic sequence, which included all the IS6 family transposases (Table 3, gene name mll6359) and multiple invasion plasmid antigens (Table 3, gene name ipaH2.5).
Virulence Factor Distribution by Function Based on Literature Search
Virulence factor synteny comparison
Since zinc binding alcohol dehydrogenases and anthranilate phosphoribosyltransferases were among the virulence factors present in the most intact prophages, we generated synteny maps for each virulence factor comparing the orientation of both in their respective prophage genomes. The synteny map of zinc binding alcohol dehydrogenase is shown in Figure 2, whereas that of anthranilate phosphoribosyltransferase is presented in Supplementary Figure S1.
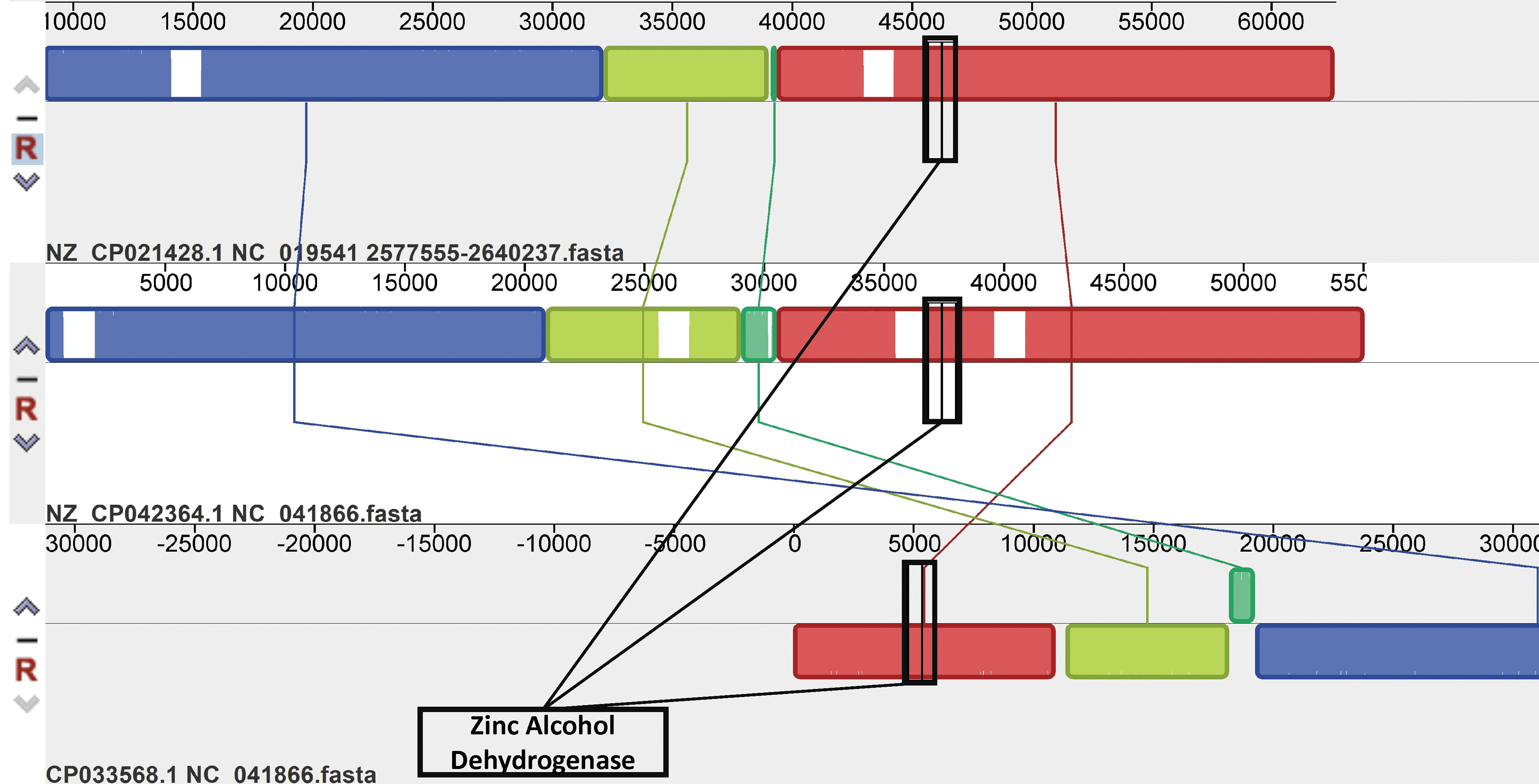
Synteny map comparing conserved alcohol dehydrogenase within Acinetobacter pittii prophages. A MAUVE synteny map was generated to analyze the zinc binding alcohol dehydrogenase genes (adhD) present in A. pittii prophages. The map compares the three prophages that possess adhD by looking at the complete nucleotide sequence of each prophage. Each colored block represents a segment of genomic material conserved across sequences, and the height of the colored similarity profile within each block correlates with the degree of conservation between sequences. 18 The virulence factor—zinc binding alcohol dehydrogenase—is present in an area of high similarity profile height (boxed in black), which suggests that the virulence factor is conserved. Genomic rearrangement of the prophage DNA occurred in the vicinity of the virulence factor, with the aquamarine block undergoing significant deletion and inversion relative to the light green block in some sequences.
In Figure 2, the virulence factor—zinc binding alcohol dehydrogenase—is present in the red synteny block for each of the three prophages. Other regions of these prophages are also highly conserved, with each homologous region delineated by a distinctly colored block. However, it can be seen that genomic rearrangement of the prophage DNA occurred in the vicinity of the virulence factors.
In the Acinetobacter phage YMC/09/02/B1251 (Fig. 2, top), the aquamarine block seems to have undergone deletion in comparison with the aquamarine blocks of the two Acinetobacter phage YMC11/11/R3177 (Fig. 2, center and bottom). In addition, the bottom Acinetobacter phage YMC11/11/R3177 possesses a large inversion of the light green and aquamarine blocks relative to the other two prophages. This change, thus, suggests that prophages and their virulence factors may have influences on genomic rearrangements in the bacterial genome.
Discussion
A. pittii is a bacterial pathogen that has the ability to cause nosocomial infections. 2 It belongs to the Acinetobacter calcoaceticus-baumannii complex, which consists of four similar bacterial species: A. pittii, A. baumannii, A. calcoaceticus, and A. nosocomialis. 25 Other species in this complex have been found to have virulence-enhancing prophages,11,12 but A. pittii has yet to be extensively analyzed for prophage sequences. Thus, this study probes and analyzes the prophages in A. pittii, as well as their effects on the bacteria's virulence.
Both A. baumannii and A. nosocomialis, the other two species in the A. calcoaceticus-baumannii complex that can cause clinical infection, 26 have been shown to possess virulence-strengthening prophages.11,12,27 As a result, the observation of a similar trend in A. pittii suggests a similar mechanism for virulence in the complex.
The A. pittii C54 strain contained a plasmid (Genbank accession: NZ_CP042365.1) that harbored a Burkholderia phage phiE12-2. This phage contained almost a fifth of all virulence factors within A. pittii prophages, as well as almost all of the antibiotic resistance genes present with high confidence within prophages. Given that A. pittii has been shown to harbor R (resistance) plasmids,28,29 it is possible that genetic recombination and rearrangement within the C54 plasmid led to the insertion of a larger amount of bacterial virulence genes into the Burkholderia phage phiE12-2 compared with other prophages in the study. If this is the case, then the C54 plasmid should be studied as a potentially novel R plasmid.
We identified a general effect of prophages in bacterial genomes that could also be linked to their virulence genes. Genomic rearrangement of prophage sequences was observed in proximity to the virulence factor zinc binding alcohol dehydrogenase. Although the influence of the alcohol dehydrogenase virulence factors on these genomic changes is unclear, the presence of such changes itself could provide evidence for the role of prophages in mediating inversions and other rearrangements in bacterial genomes. 30 The increased genetic diversity could aid bacteria in adapting to stressful environments during infection.
Two prophages were identified in the study, with both being prevalent within A. pittii and containing a large quantity of virulence factors: Acinetobacter phages YMC11/11/R3177 (Genbank accession NC_041866.1) and YMC/09/02/B1251_ABA_BP (Genbank accession NC_019541.1). The former has not been extensively researched, whereas studies on the latter have demonstrated that YMC/09/02/B1251_ABA_BP is a ubiquitous and mobile prophage shared among many A. baumannii strains. 31 The similarity between the A. pittii and A. baumannii prophages is shown in Supplementary Figure S2. As a result, these two prophages could be major mediators of virulence gene transfer within and between Acinetobacter species, and it is necessary that they be studied further.
This study provides conclusive evidence for the existence of virulent prophages within A. pittii. However, it was limited by a few factors. The sample of 25 A. pittii strains used in the study was relatively small, as only these strains were available at the time of the study. The small sample size could have also affected our search for the most prophage-dense regions in A. pittii chromosomes. This study focuses on a bioinformatics approach to analyze the bacterial genomes, and the evidence provided is not supported by experimental data.
Given A. pittii's increasing clinical relevance, it is expected that more strains will be catalogued in the future. This will allow subsequent A. pittii analysis to be more comprehensive and revealing and an opportunity to experimentally test some of the predictions from our study. In addition, our study was stringent in applying high cutoff standards: We analyzed only intact prophages and virulence genes that were present with an E-value of less than 1 × 10−20 when compared with VFDB. This allowed a high-degree confidence in our results.
In general, our study revealed the existence of numerous prophages within A. pittii. We catalogued these sequences' effects on bacterial virulence, antibiotic resistance, and genomic rearrangements. Our results further the understanding of this nosocomial pathogen, its pathogenicity mechanisms, and its bacterial relatives.
Conclusion
A. pittii, a nosocomial pathogen, contains prophages that could impact its virulence, antibiotic resistance, and cause genomic rearrangements. The prophage sequences contain many virulence factors and antibiotic resistance genes, some of which—such as beta lactamases—mirror existing resistance phenotypes in A. pittii. This provides evidence for prophages serving as current and future influences on the bacteria's pathogenicity mechanisms.
Moreover, the results demonstrate that A. pittii and other members of the A. calcoaceticus-baumannii complex exhibit similar patterns in terms of the spatial distribution of prophages and the prevalence of common prophages (i.e., YMC/09/02/B1251_ABA_BP). Further analysis of common virulence and prophage trends in A. pittii and other members of the complex is warranted. Such research will help illuminate the extent to which prophages have and continue to influence the pathogenic phenotypes of bacteria, which could have clinical ramifications.
Footnotes
Authors' Contributions
All people who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the article. Further, each author certifies that this material or similar material has not been and will not be submitted to or published in any other publication before its appearance in the Phage: Therapy, Applications, and Research journal. R.Z. conceived of the project idea, drafted the methodology, and performed the data analysis. V.M. and R.Z. worked together to interpret the analysis results, after which R.Z. wrote the article. V.M. provided general structural guidance during the article-writing process. V.M. also supervised the project during its entire duration.
Acknowledgment
The authors would like to thank Dr. Anne Rosenwald and Dr. Gaurav Arora for providing feedback on this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.