
Abstract
Background:
The experimental determination of a bacteriophage host is a laborious procedure. Thus, there is a pressing need for reliable computational predictions of bacteriophage hosts.
Materials and Methods:
We developed the program vHULK for phage host prediction based on 9504 phage genome features, which consider alignment significance scores between predicted proteins and a curated database of viral protein families. The features were fed to a neural network, and two models were trained to predict 77 host genera and 118 host species.
Results:
In controlled random test sets with 90% redundancy reduction in terms of protein similarity, vHULK obtained on average 83% precision and 79% recall at the genus level, and 71% precision and 67% recall at the species level. The performance of vHULK was compared against three other tools on a test data set with 2153 phage genomes. On this data set, vHULK achieved better performance at both the genus and the species levels than the other tools.
Conclusions:
Our results suggest that vHULK represents an advance on the state of art in phage host prediction.
Introduction
Bacteriophages (or simply phages) are believed to be the most diverse and abundant biological entities in the biosphere. 1 They are present in any place or environment as long as a prokaryotic host is nearby. Recent improvements in next generation sequencing techniques and the rise of metagenomics have generated large and diverse collections of viral genomes. Thousands of DNA sequence data sets from different types of environmental or host-associated samples, such as ocean, soil, and animal guts, have become available,2–4 thus shedding light on the so-called viral dark matter.5–7 Although phage genomes are being sampled at a rapid pace,6,8 acquisition of new knowledge about phages has been restricted by our capacity of isolating them through cultivation-based methods. 9
Phages regained prominence in the past few years, thanks to phage therapy, which is considered as a promising approach against multidrug-resistant bacteria. 10 A key step in phage therapy development is finding a phage that can infect and efficiently terminate with high specificity an infectious agent of interest. Establishing a link between phage and host is, however, far from trivial, since phages may present a variety of infecting molecular mechanisms, and a host range that varies from narrow (strain-specific) to very broad (hosts from a same genus, family, or higher taxonomic levels).11–13
Furthermore, host range characterization mostly depends on laborious wet-laboratory experimental methods and may be very specific for species or even bacterial strains. 9 Because of the laboriousness and specificity of these methods, effective computational prediction of phage hosts can potentially be a time- and labor-saving approach.
Edwards et al 14 have reviewed genomic signals used to link a phage to its host. Oligonucleotide frequency profiles, sequence similarity, and CRISPR spacers matches were evaluated. Sequence similarity between phage and host genome and matches of CRISPR spacers were found to be more effective in establishing phage–host pairs. However, only 40% accuracy was achieved with these features. Broader approaches have been suggested, such as the use of Markov models based on k-mer frequencies, implemented in the WIsH tool, 15 which showed 63% of mean accuracy when predicting host genus among 20 possible genera.
A recent study proposed the use of neural networks based on an ensemble of distance measurements and other features, concepts that were implemented in the tool VirHostMatcher-net (VHM-net). 16 VHM-net achieved accuracies that varied from 43% to 59% at the host genus level only and is theoretically able to predict thousands of different prokaryotic hosts. The tool RaFAH uses a machine learning approach based on protein clusters to assign hosts to complete or fragmented genomes of prokaryotic viruses, with a performance comparable with that of other methods for virus–host prediction. 17
Young et al have recently evaluated different levels of genomic features for phage host prediction. 18 These authors defined four levels of features that could be extracted from genomes by applying different annotation processes: oligonucleotide frequencies, amino acid frequencies, protein physicochemical properties, and protein domains. They conclude that all feature levels yield significant signals for host prediction, probably being complementary to one another.
In this study, we present a novel neural network-based tool for phage host prediction based on annotated genomic features (protein-coding genes). Features are extracted from phage genomes and searched against a protein database, which provides information about genes and their function. These features are converted to a vector with these features and fed to multilayer perceptron (MLP) models trained to predict 118 and 77 different host species and host genera, respectively. Trained models were implemented in a user-friendly tool named vHULK, which achieved better accuracy than competing tools in our tests.
Materials and Methods
Data sets
The GenBank nucleotide database of viral records was downloaded in April 2020. Records passed through a quality control of available metadata, and 8616 records were selected to create the baseline data set. To remove redundancy in terms of presence of similar genomes in the baseline data set, we calculated a pairwise matrix of Amino Acid Identity (AAI) scores using the CompareM AAI_wf tool (https://github.com/dparks1134/CompareM).
We parsed AAI scores matrices to generate three AAI redundancy reduction data sets for each prediction type (host species and host genus) as follows: Instances with AAI scores equal to or higher than 90%/80%/70% were removed and only one randomly chosen instance from each group was left in the AAI redundancy reduction data set, for the clusters in which all members had the same host. For clusters in which more than one host was annotated, we kept one genome per host. Each AAI redundancy reduction data set was randomly split into training (for neural network fine-tuning purposes) and test sets in a 70/30 manner, that is, 70% of the data set was used to train the neural network and 30% to evaluate its predictions. Performance measurements were averaged for 10 random splits of the training/test data sets.
Two neural network predictors were trained on the baseline data set: one for prediction of host species and another for prediction of host genus. Each of these models required its own training set; we call them the species training set and the genus training set. For adequate model training, for each host species or host genus to be predicted (i.e., to define a target), we needed minimum number of training instances to avoid overfitting. Thus, for a given species or genus to be included as a target, we required the existence of at least six phage records with that host. As a result, the data set for training the genus model contains 77 targets and 7831 instances (phage genomes), and the species model contains 118 targets for prediction and 6922 instances (Table 1). These data sets were used for final model training.
Target Hosts and Number of Instances in the Final Training Set for Each Model Type (Species and Genus)
As an approximation of a real-life use case, we created the newly deposited genomes (NDG) data set. This data set includes GenBank records released between April 1, 2019, and March 31, 2020; these records were not part of the training sets. The Quality Control filters applied to generate the baseline data set were applied, and only complete genomes from viruses infecting Bacteria and Archaea hosts were considered (including host targets that were not present in the training sets). A total of 2153 genomes met these criteria.
Feature engineering and representation
Genomes in the training species/genus data sets were individually submitted to Prokka 19 for automatic gene prediction and annotation. Predicted protein sequences (FASTA amino acid format) were searched against the pVOGs database of phage protein families 20 using HMMscan. 21 Search results were parsed to populate a matrix of pVOG identifiers in columns and phage genes in rows. The matrix cell values were based on the mathematical sigmoid transformation of the e-value of the best alignment of each gene against the pVOGs database. The best alignment e-value is transformed by a function that is the sigmoid of the negative logarithm of the e-value, which is then multiplied by a factor of 10 for better scaling.
With this approach, each genome is represented by a matrix of numbers between 0 and 10, and values close to 10 mean that significant hits regarding specific phage protein families are present among phage genes for that genome. Then these matrices were transformed to vectors by adding up column values, making the data representation easier for neural network interpretation and training. Since the data set is unbalanced with respect to representation of its classes, an importance weight was implemented for each class during training to account for the imbalance.
Machine learning model architecture, configuration, and training
The model for genus prediction is MLP 22 constituted of one input layer with dropout rate of 35%, two hidden layers (with, respectively, 1000 and 300 neuron units, with SELU activation function), and one output layer, with Softmax activation function. Optimization in training was made with the ADAM algorithm. 23 Categorical cross-entropy was used as a loss function with Early Stopping implemented. Regularization L2 was used in the first neural layer.
Each genus score has an entropy value and an energy level associated with it (more on this is given hereunder), which may be used to gauge the uncertainty of each score.
Final models were trained with the 90% AAI redundancy reduction species/genus data sets and saved with the format TensorFlow SavedModel. Training code was written in Python 3.8.5 using TensorFlow 2.9.1, Keras 2.9.0, and KerasTuner 1.1.2 libraries. Training was performed in a server equipped with an Nvidia GPU Quadros P6000 processor.
Tool implementation
Models for prediction of host species and genus were embedded in a standalone toolkit developed in Python 3, named vHULK, for Viral Host UnveiLing Kit. The tool accepts as input phage genomes FASTA files and outputs top prediction scores for each internal model. To complete the toolkit, vHULK also outputs an entropy value and an energy level for the vector of prediction scores of the genus model, which will provide measures of prediction confidence. With the tests made with the NDG data set, we concluded that for genus predictions entropy values and energy could inform confidence of the predictions (Table 2). For species predictions we were unable to use entropy and energy values to provide confidence scores.
Relation of Energy and Entropy Values of Scores and Confidence of the Genus Predictions
Comparison of vHULK with VHM-net, CRISPR spacers matches, and RaFAH
Phage genomes of the NDG data set were submitted for host prediction using four different approaches: vHULK, neural networks with distance measurements, and other features used by VHM-net, 16 RaFAH, 17 and CRISPR spacers as links between host and phage genomes. Default parameters and databases were used for vHULK, VHM-net, and RaFAH. For the CRISPR spacers approach, individual genomes were searched against a database of CRISPR spacers of the CRISPRCasDB 24 using the NCBI blastn tool. 25
To adapt our search to CRISPR spacers characteristics, word size was set to 9 nt, and thresholds of e-value ≤0.001 and number of mismatches ≤2 were applied to select significant hits. Only the best scoring hit was considered in each approach. In all four cases, predicted hosts were compared with known phage hosts as specified by the tag “host” or “lab_host” in the GenBank record.
Results
Phage–host prediction features
A total of 9504 features were generated to train the vHULK neural network models in predicting phage hosts. Features are transformed e-values obtained from similarity searches to a corresponding prokaryotic Virus Orthologous Group (pVOG) (Fig. 1). 20
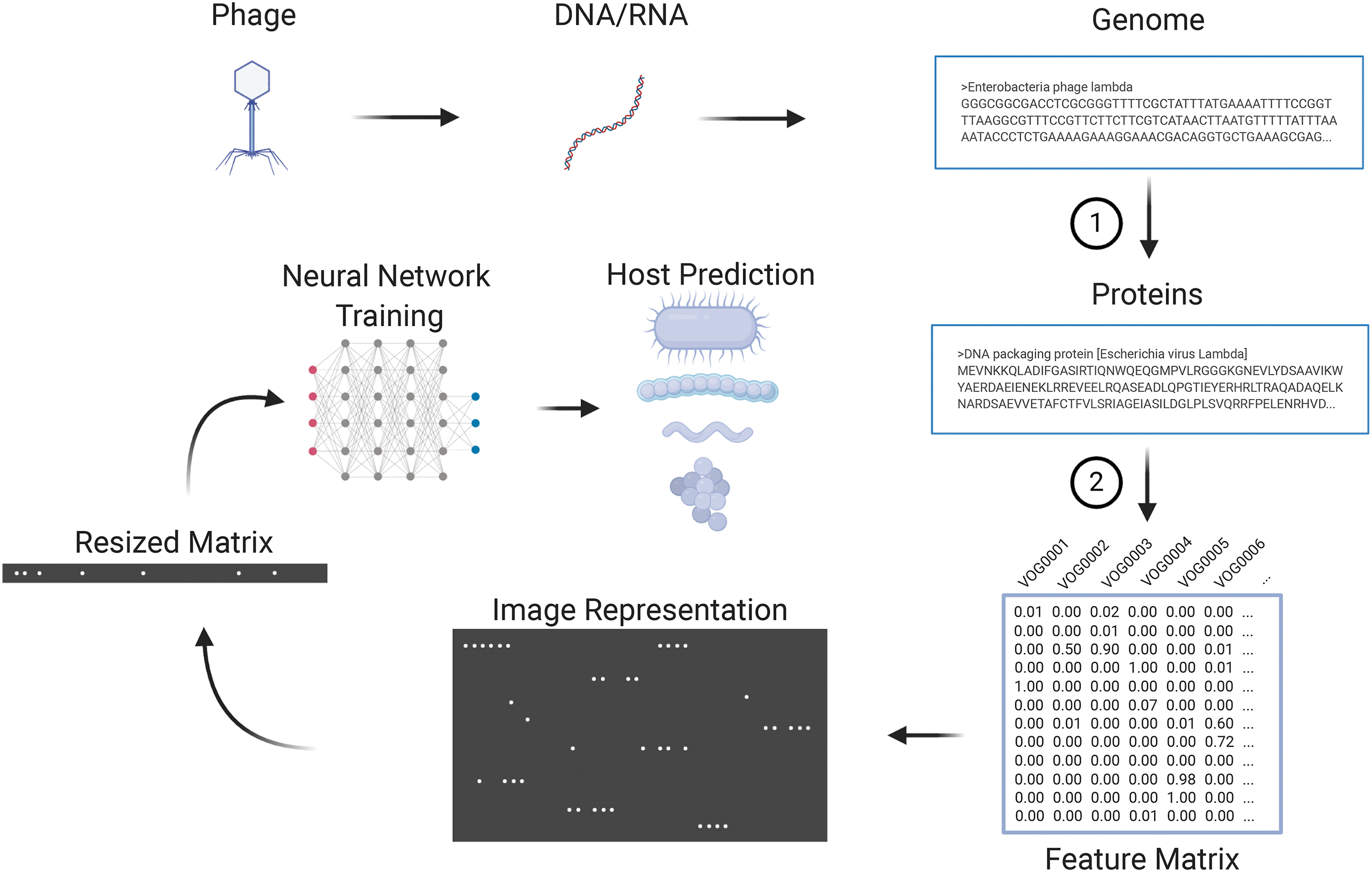
Pipeline for phage prediction. Schematic representation of how high-level features are engineered to feed the neural network and to perform host prediction. (1) Gene prediction and annotation by Prokka; (2) HMMscan search against pVOGs database of phage protein families and generation of a matrix with transformed e-values. pVOG, prokaryotic Virus Orthologous Group.
We evaluated feature relevance by using a predominant correlation approach as implemented in the Fast Correlation-Based Filter algorithm. 26 In this way we identified a set of features that seem to be strongly correlated with host prediction in specific groups of phages. Among these, features that represent the orthologous groups VOG4545 and VOG4558 had two of the highest correlation values. VOG4545 is an orthologous group ubiquitous among the 3 tailed phage families (Myoviridae, Siphoviridae, and Podoviridae), composed of 1747 known orthologous proteins. Most of the VOG4545 member proteins are annotated as a tape measure protein, which is thought to vary in length according to phage tail size. 27
Proteins of the orthologous group VOG4558 are present in phages of the Siphoviridae family, mainly infecting hosts of the genera Mycobacterium and Gordonia. They are annotated as a structural minor tail protein. It is worth noting that pVOG annotations of most of the top correlated features indicate proteins directly or indirectly related to the infection mechanism. Unweighted and weighted information theory metrics for all features used in this study were obtained (Supplementary Table S1).
Neural network testing on the AAI redundancy reduction data sets
vHULK was run on the AAI redundancy reduction data sets (Table 3). F1-scores ranged from 63% to 80% for genus predictions, whereas species predictions resulted in F1-scores ranging from 43% to 68%. Macro versions of the F1-score resulted in lower values (40–55% at genus level and 57–64% at the species level).
Performance Results (in %) on the AAI Redundancy Reduction Data Sets Using Different Levels of Redundancy Removal
Values represent the average of 10 random splits, and standard deviations are shown in parentheses.
AAI, Amino Acid Identity.
We obtained the confusion matrix for predictions at the genus level using the 90% AAI redundancy reduction data set (Fig. 2). As expected, most of the predictions are centered around the diagonal line. Most of the confusion happened with hosts from the same taxonomic family. These results are expected and may be biologically meaningful, since wide host range phages have been reported to infect species belonging to different genera from the same family.28,29 We have generated detailed confusion matrices showing average percentage of actual classes for five random split train/test data sets (Supplementary Table S2).
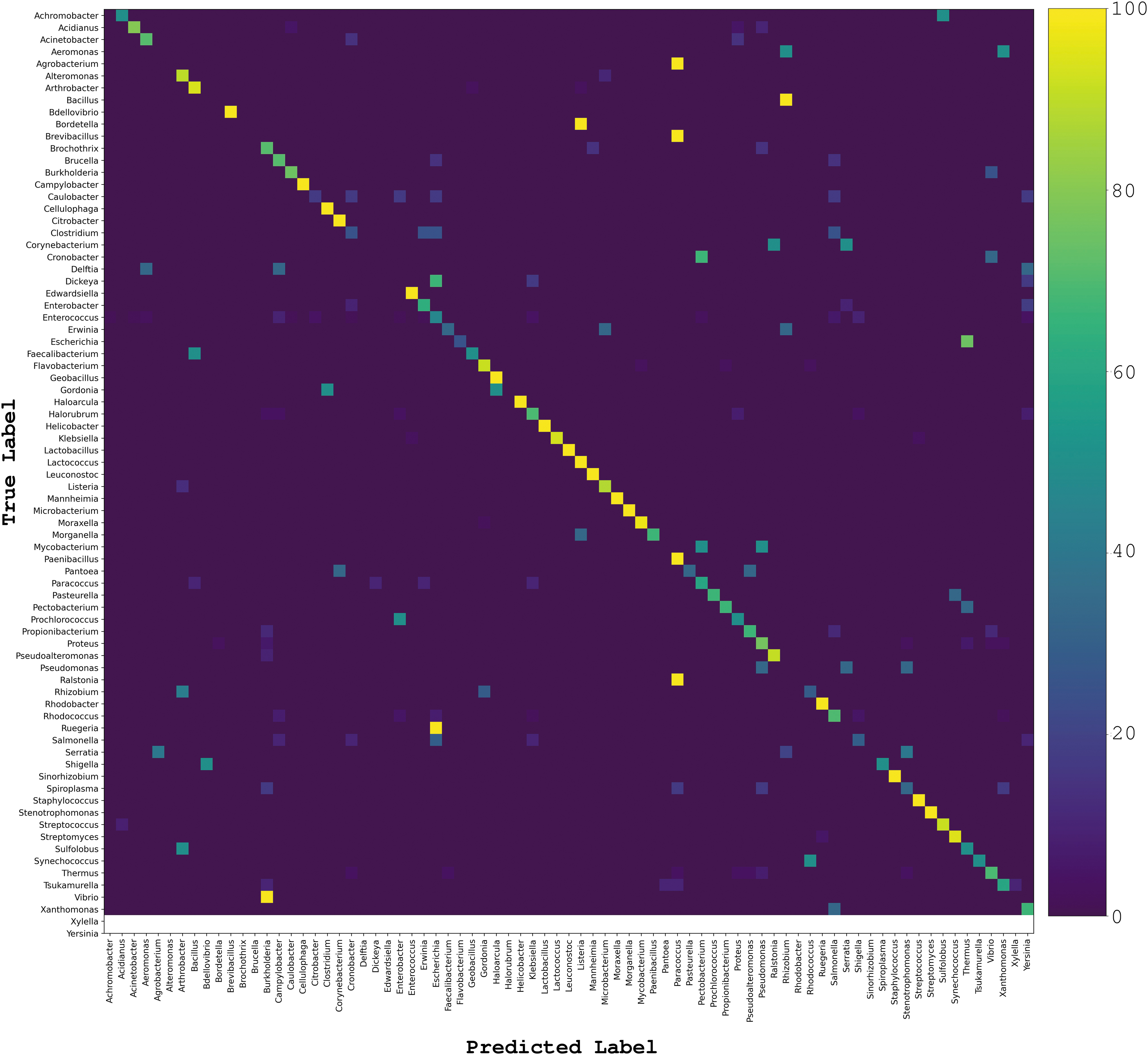
Confusion matrix of host predictions. Confusion matrix for predictions at the genus level using the 90% AAI redundancy reduction data set. Color scale corresponds to average percentage values of five random test splits. Corrected predictions are centered on the diagonal line. AAI, Amino Acid Identity.
Performance results on the NDG data set
We used the NDG data set as an approximation of a real-life use case (see Materials and Methods section). Performance on the NDG data set varied depending on the host and type of prediction (genus or species). We have obtained performance results for hosts of the ESKAPE group, which are bacteria known to be multidrug resistant (Table 4). Complete descriptions of vHULK output results as well as performance metrics for all phages of the NDG data set are available (Supplementary Tables S3–S5). Density distribution of entropy and energy levels of the predictions on the NDG data set are available (Supplementary Fig. S1).
Precision/Recall Details for Six Bacterial Hosts of the ESKAPE Group
Total counts of genomes in the NDG data set for each host are shown, as well as precision/recall percentages. E. faecium is not one of the possible hosts in vHULK's predictions; therefore, predictions are shown at the genus level only.
NDG, newly deposited genomes.
Comparing phage host prediction programs
We have compared vHULK host predictions with those provided by VHM-net, RaFAH, and CRISPR spacers matches; the NDG data set was used for this purpose (Fig. 3). F1-score for vHULK predictions was three times higher than the second-best approach, when predictions at the species level are considered (44% against 13% of CRISPR spacers). When predictions at the genus level are considered, vHULK obtained an F1-score of 79% across 77 possible host targets, whereas RaFAH (the second best) obtained 65%. We provide detailed information about predictions in the NDG data set for the four methods (Supplementary Table S6).
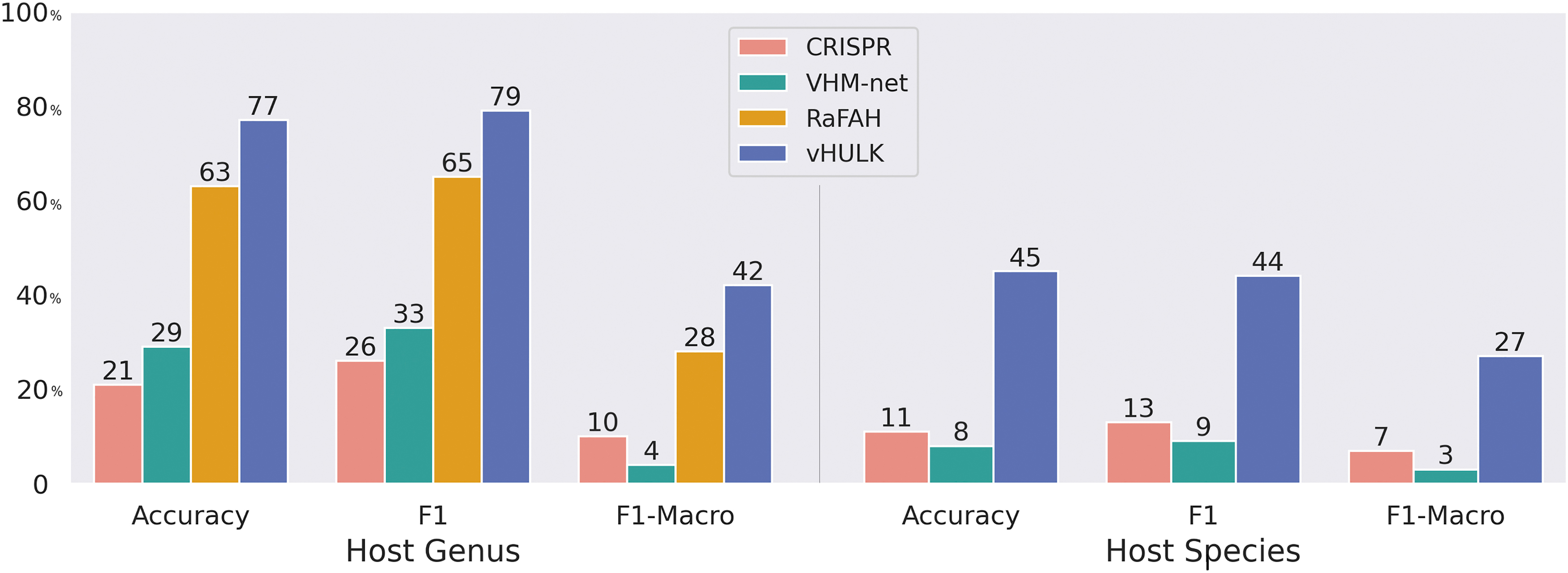
Performance metrics of vHULK, VirHostMatcher-net RaFAH tools, as well as the CRISPR spacers matches approach. Predictions were obtained for 2153 phage genomes of the NDG data set, both at the species and genus levels. NDG, newly deposited genomes.
All 4 programs ran on a server with Intel Xeon 2 GHz CPUs, 22 threads and 32 GB of RAM. vHULK took ∼18 h of wall time, VHM-net took ∼32 h, CRISPR spacers took ∼50 min, and RaFAH took ∼15 h. Genomes were submitted sequentially, and no additional parallelization was used in any of the tools.
Discussion
We have presented vHULK, a new program to predict phage hosts based on phage genome sequences. A comparison between vHULK and three other tools/methods for host prediction showed that our program had superior performance at both the species and genus levels, and in all three main metrics considered (Accuracy, F1, and F1-Macro).
Testing of vHULK on data sets with decreased protein sequence redundancy levels showed F1-scores that varied from 68% to 43% at the species level and 80% to 63% at the genus level. These results show clearly that the less redundant a data set is in terms of protein similarity, the lower the scores achieved by vHULK. This shows the dependency that our tool has on protein sequence similarities between training data and test data, as one would expect, given the tool's design. Nevertheless, the results suggest that the tool can perform fairly well even with the lack of redundancy.
The species model performs very well when compared with other tools. Although it is a major improvement, this level of taxonomic prediction still needs more work for robust usage.
vHULK was trained to predict 118 different species and 77 different genera. These number of possible hosts were limited only by the number of instances available to train the model and by our requirement that there had to be at least six records for any given species or genus. In total, 118 and 77 are tiny numbers when compared with the presumed richness of phage hosts that exist in the biosphere. We expect, however, that the number of species and genus hosts in the vHULK repertoire will grow with future releases of the software, based on the growing number of available phage and bacterial genome sequences, and lab-tested information on phage hosts. 30
Nevertheless, despite the relatively small host repertoire, we envision that vHULK will grow as a utility for researchers as the tool is updated and grows in repertoire. One use could be the mining of metagenomic data sets for new phages that may have as host one of the six multidrug-resistant bacterial pathogens that compose the ESKAPE group (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.). 31 All of them are part of the vHULK host repertoire.
It is known that many machine learning prediction models may struggle when facing instances for which there was no target in training.15,16,18 We have tried to mitigate this problem by providing users with entropy and energy values associated with scores; we have shown that these can be used as proxies for prediction confidence.
Regarding the design of vHULK, we would like to point out that the program embodies a novel approach for extracting and representing genomic features for use in neural networks. The main novelty is the use of alignment significance scores when evaluating presence/absence of protein families in phage genomes, and their number when they are present, as features in the neural network. The protein features in vHULK are somewhat related to the protein domains of Young et al. 18 However, whereas they proposed a count of domains, we use alignment significance scores. Our results demonstrate that the use of these features was a determining factor in achieving good accuracy. We conjecture that this same approach can be extended to the prediction of other viral biological attributes, such as viral life cycle and virulence.
Conclusions
Our results show that vHULK achieves the best performance among existing programs for phage host prediction, and should, therefore, be a useful tool to the scientific community interested in such predictions.
Footnotes
Acknowledgments
The Nvidia GPU Quadro P6000 unit used in this study was obtained through a donation by the Nvidia Corporation to J.C.S.
An earlier draft of this study was posted as a preprint at bioRxiv (DOI: 10.1101/2020.12.06.413476v1.full.pdf?). This study was also based in part on Deyvid Amgarten's research for his doctoral thesis (available at: ![]() ).
).
Authors' Contributions
All authors have read and approved this article.
Software Availability
The tool, the models, and the testing examples and documentation are publicly available in GitHub: (https://github.com/LaboratorioBioinformatica/vHULK). We also provide GenBank records used to train vHULK's models and accessions numbers for the NDG data set in (![]() ).
).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded in part by the São Paulo Research Foundation (FAPESP) (Grant No. 11/50870-6) and CAPES Grant No. 3385/2013. Deyvid Amgarten and Bruno Koshin Vázquez Iha were funded in part by student fellowships from CAPES. Aline Maria da Silva and João Carlos Setubal were funded in part by senior research fellowships from the National Council for Scientific and Technological Development (CNPq).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.