
Abstract
Background:
Aeromonas hydrophila is a prevalent pathogenic bacterium in aquaculture that causes economic loss around the world. Antimicrobials are used to control and prevent the incidence of bacterial pathogens in aquaculture. However, they lead to the emergence of antimicrobial resistance strains and the accumulation of antibiotic residues in fish tissue. To address these issues, bacteriophages may be promising alternatives to many antibiotics in combating bacterial infections in aquaculture.
Materials and Methods:
The phage specific to A. hydrophila was isolated from domestic wastewater. The morphology of phages was analyzed using transmission electron microscopy. The genomic DNA of the Aeromonas phage T65 strain (APT65) phage was sequenced with a paired-end read length of 2 × 150 bp. The genome sequence was assembled and annotated. The tRNAs were predicted, and antimicrobial resistance and virulence genes were screened. A representation of the APT65 genome was constructed.
Results:
The genome of APT65 is linear double-stranded DNA with 85188 base pairs having 116 open reading frames (ORFs) and a G + C content of 39.41%. The 32 ORFs were predicted to encode proteins with known phage functions. No virulence factors, antibiotic resistance genes, or temperate lifestyle genes were found. The phage is icosahedral and measures 60 nm in diameter. Based on the whole genome sequence, APT65 belongs to Lahexavirus.
Conclusions:
The taxonomic analysis of the phage with a genome length of 85,188 bp revealed that it is a new species of the genus Lahexavirus. We announce the whole genome sequence of APT65, which should be named Lahexavirus APT65, as well as the absence of antimicrobial resistance and virulence factors from its genome. Based on our results, the Lahexavirus APT65 phage may have potential as a therapeutic agent to tackle antimicrobial resistance in aquaculture.
Introduction
Intensive aquaculture increases the risk of infection by opportunistic pathogens, thus resulting in dependence on antimicrobial agents. 1 Aeromonas hydrophila, an opportunistic aquatic pathogen, is the most prevalent cause of motile Aeromonas septicemia (MAS) in many fish species living in fresh and brackish water.2,3 MAS has resulted in substantial economic losses for the aquaculture sector throughout the globe. 4 Also poikilothermy species, such as amphibians, fish, snakes, and turtles, are among the most susceptible to A. hydrophila infection. 5 It is a zoonotic pathogen that causes gastroenteritis and other systemic diseases in humans and other species. 6
When the water quality parameters are unsuitable for fish, A. hydrophila may induce mass mortality. 7 Antimicrobials are used to prevent mortality, which triggers the emergence of antimicrobial resistance bacteria and the accumulation of residues in fish tissue.8,9 Antimicrobial resistant strains are able to reproduce and transmit their antibiotic resistance, resulting in the emergence of a large number of antibiotic-resistant bacteria. 10 To overcome these issues, bacteriophages can be used as an alternative to many antibiotics to control bacterial infections in aquaculture. 11
The quick generation time, nontoxicity, high specificity to the bacterial host, and the fact that they do not negatively impact the viability of other flora in the environment make bacteriophages a promising, environmentally friendly, and sustainable antibiotic alternative in aquaculture. 12 Considering the advantages of phages, a novel bacteriophage, Aeromonas phage T65 strain (APT65), specific to A. hydrophila, was isolated from domestic wastewater, 11 and the whole genome of APT65 was mapped out in this study.
Materials and Methods
The phage specific to A. hydrophila was isolated from domestic wastewater in Trabzon, Türkiye. Thirty milliliters of filtered wastewater was mixed with 30 mL of tryptic soy broth that contained 1 mL of A. hydrophila and incubated at 25°C for 24 h. The sample was centrifuged for 15 min at 5000 × g, and the supernatant was filtered through a 0.22 μm syringe filter. After repeating this procedure three times, the double-layer agar technique was employed to detect phage activity.11,13 The characteristics of the APT65 were described in the earlier work done by Ture et al. 11 Initial concentration of the phage was 2 × 109 PFU/mL.
The morphology of the phage was analyzed using transmission electron microscopy on negatively stained specimens. In brief, the phage solution was centrifuged for 2 h at 41,000g, and the pellet was resuspended at 1010 PFU/mL in the saline magnesium buffer (0.1 M NaCl, 8 mM MgSO4, 7H2O, 50 mM Tris-HCl pH 7.5, %1 gelatin). The 10 μL of the concentrated phage solution was adsorbed on the carbon-coated matrix and the sample was finally stained with 1% uranyl acetate for 15 s. The grid was examined with a transmission electron microscope after being rinsed twice with ddH2O and air dried before examination.
Genomic DNA of APT65 was extracted using the phenol-chloroform-isoamyl alcohol procedure. 11 The sequencing library was prepared using the NEBNext DNA Library Prep Kit (New England Biolabs) and sequenced on the Illumina HiSeq4000 platform with a paired-end read length of 2 × 150 bp. The genome sequence was assembled using SPAdes 3.13.0 and annotated with Prokka 1.14.6, using the default settings.14,15 Further annotation was conducted by BLASTp using the nonredundant protein database and hidden Markov models (https://www.ebi.ac.uk/Tools/hmmer/). Then tRNAs were predicted using Aragorn v. 1.2 software 16 and antimicrobial resistance or virulence genes were screened by ABRicate. 17 Furthermore, the phage lifestyle was determined using PhageLeads to predict its suitability for phage therapy. 18 The CGView Server was used to create a depiction of the phage APT65 genome. 19
Combining phylogenetic and clustering methodologies, the Genome Blast Distance Phylogeny web application of Virus Intergenomic Distance Calculator (VIRIDIC) was used to classify APT65. The VIRIDIC approach extends and improves on the standard BLASTN method, used by the International Committee on Taxonomy of Viruses (ICTV) to determine how closely related viral genomes are and how they are linked. VIRIDIC classifies phage genomes based on a prokaryotic virus classification algorithm. 20 VirClust was used to cluster bacteriophages according to their protein profiles. 20 The nucleotide similarity of the phages was determined by using the Orthologous Average Nucleotide Identity Tool (OAT; v0.93.1) that measures overall similarity between two genome sequences. 21 The cutoff for species demarcation is 95–96%. To demonstrate that the phage is novel.
Results
The APT65 genome was 85188 bp long and contained 39.41% G+C. A total of 116 open reading frames (ORFs) ranging from 303 to 4512 bases were predicted. Among the 116 ORFs, 32 showed similarities to known phage proteins, whereas 73 were labeled as hypothetical proteins, most likely due to a lack of knowledge regarding the functional genes of Aeromonas phage genomes. Although CRISPR repeats and rRNA were not found, 12 tRNAs [tRNA-Val(tac), tRNA-Lys(ttt), tRNA-Gln(ttg), tRNA-Arg(tct), tRNA-Leu(tag), tRNA-Leu(taa), tRNA-Asp(gtc), tRNA-Pro(tgg), tRNA-Gly(tcc), tRNA-Asn(gtt), tRNA-His(gtg), and tRNA-Thr(tgt)] were identified (Fig. 1). Based on the whole genome sequence, the APT65 phage belongs to the genus Lahexavirus (GenBank accession: OP491958.1), and it should be named Lahexavirus APT65.
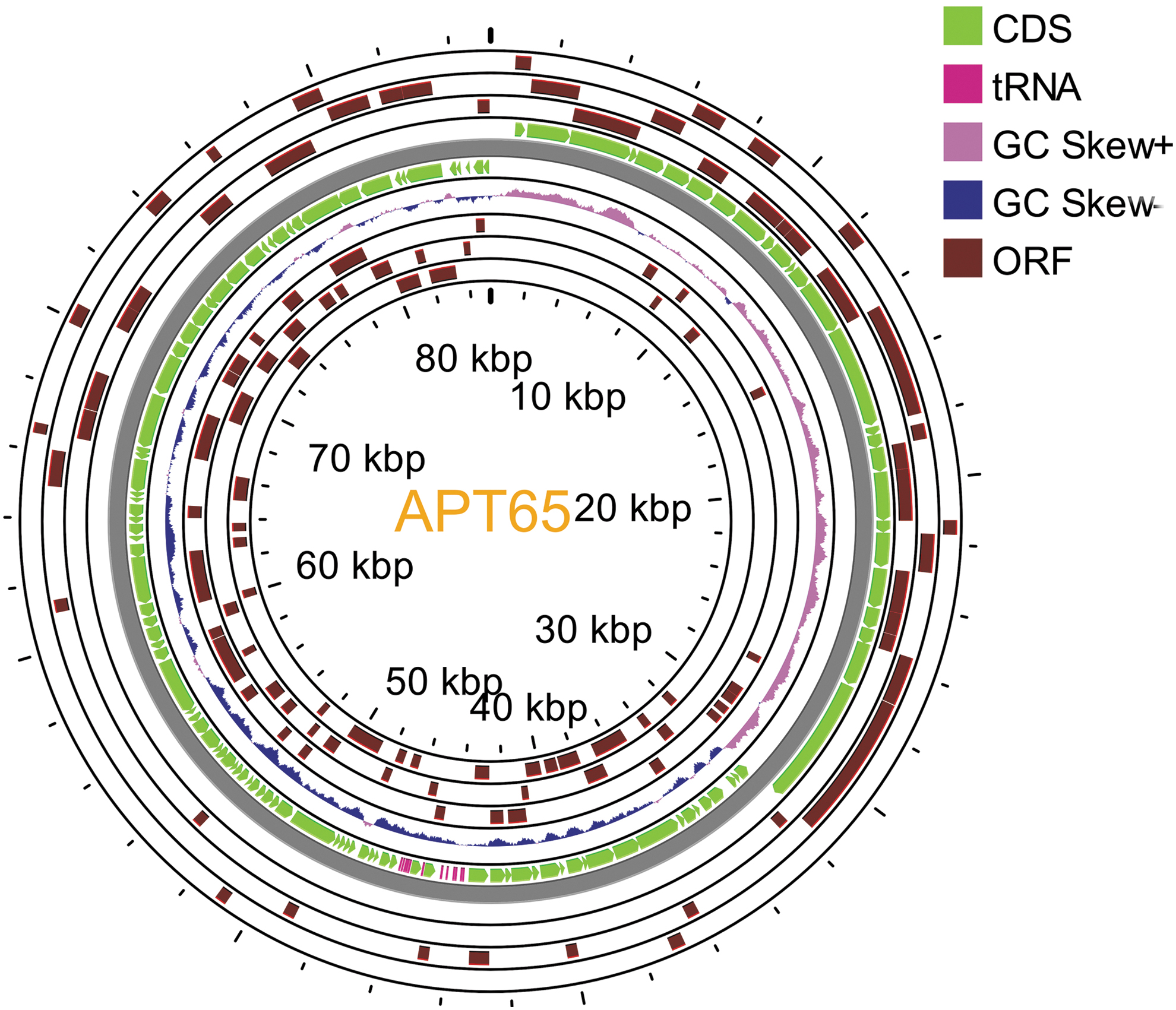
ORF map of the genome of Aeromonas phage, APT65. The genomic map was prepared by the CGView Server. The outer lane and the next lane display genes on the forward and reverse strands, respectively. APT65, Aeromonas phage T65 strain; ORF, open reading frame.
Protein profile-based phylogenetic analysis revealed that Aeromonas phage APT65 was closely related to Lahexavirus LAh6, Lahexavirus LAh8, Lahexavirus lv44572, and Lahexavirus LAh 9, but not to Siphoviridae, Haloferacalesvirus, Jhansiroadvirus, Przondovirus, Lafunavirus, or Lagaffevirus (Fig. 2). Pairwise intergenomic distances/similarities across viral genomes revealed that Lahexavirus LAh6, Lahexavirus LAh8, Lahexavirus LAh9, and Lahexavirus lv44572 had >88% similarity, whereas these phages shared between 45% and 47% similarity with the Lahexavirus APT65 phage (Fig. 3). Lahexavirus LAh9 and Lahexavirus lv44572 had 97.9% nucleotide similarity according to average nucleotide identity (ANI) results, whereas these phages shared 66.8% similarity with the APT65 phage (Fig. 4). These three phages bore no resemblance to the other phages.
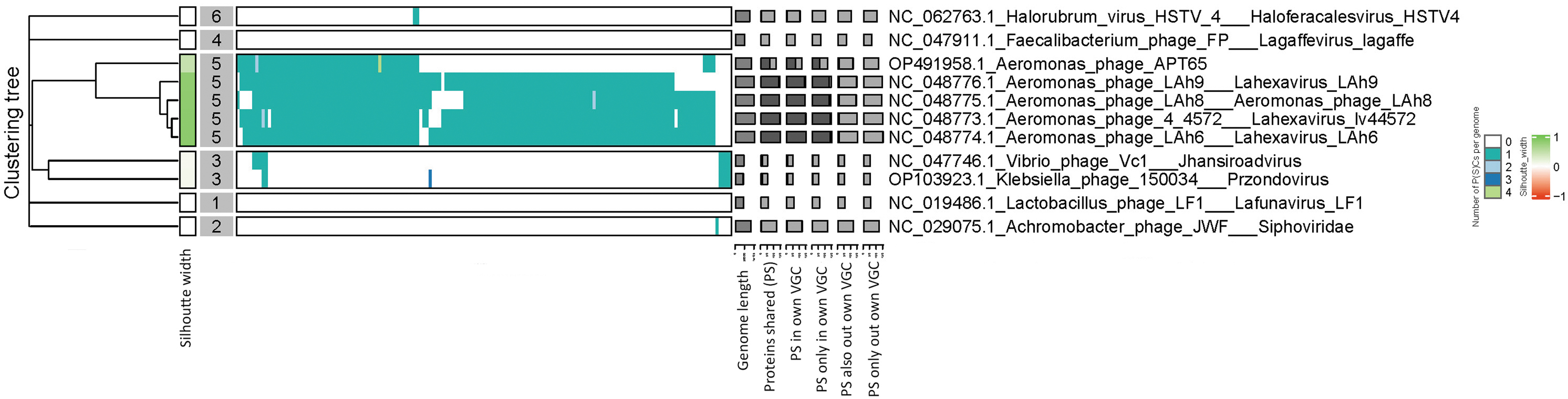
The clustering of viral bacteriophage proteins into two different levels. Proteins are first grouped into PCs based on their reciprocal BLASTP similarities; these PCs are then grouped into protein superclusters based on their HMM similarities. HMM, hidden Markov model; PCs, protein clusters.
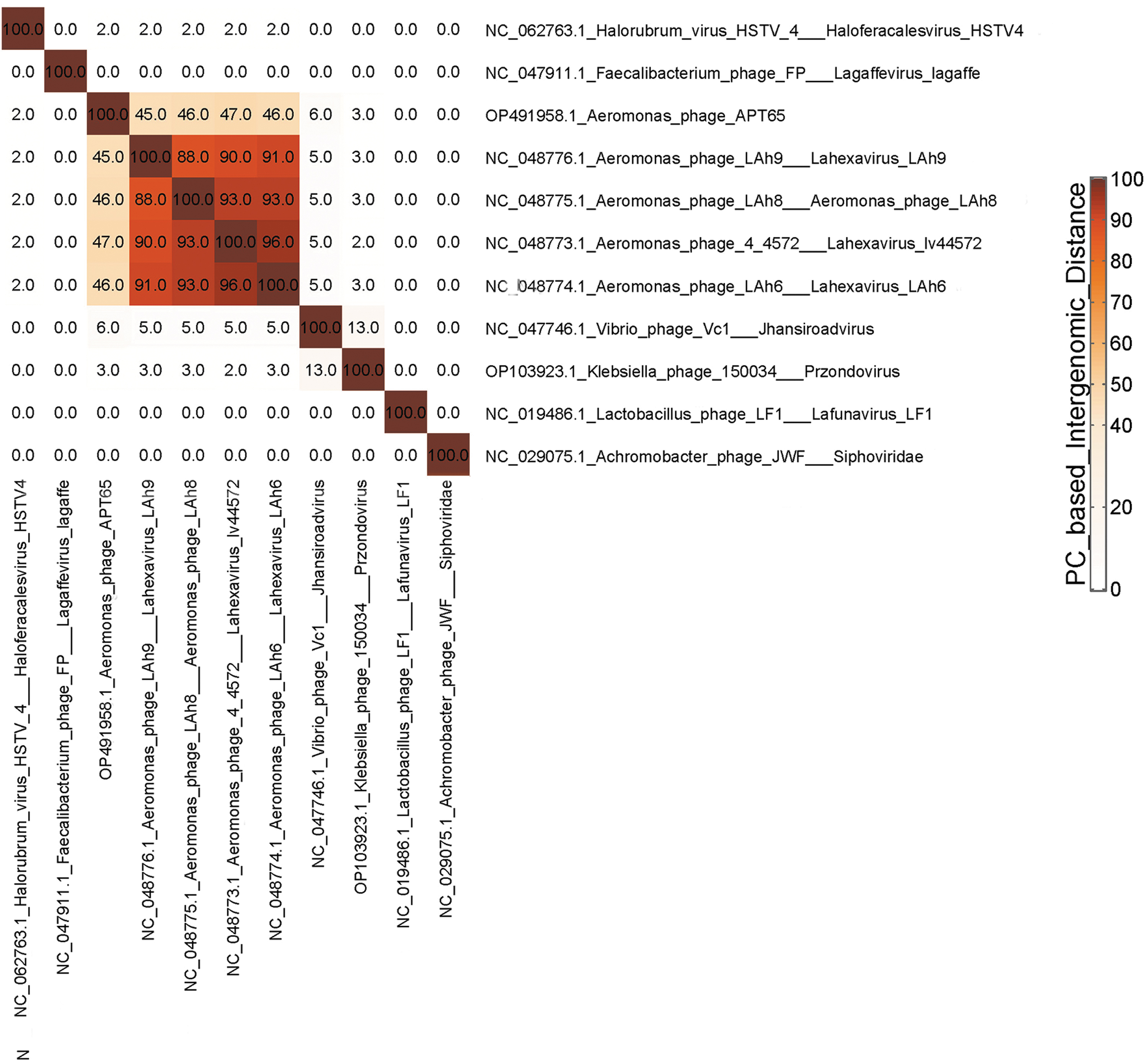
Distance Heatmap among bacteriophage genomes.

ANI value of the APT65 phage in comparison with related and unrelated bacteriophages. ANI, average nucleotide identity.
Furthermore, no virulence factors, antibiotic resistance, or temperate lifecycle genes were found. The virions were icosahedral, 60 nm in diameter, with a lengthy contractile tail of 170 nm (Fig. 5). The results show that the Lahexavirus APT65 phage could be used safely to treat or prevent A. hydrophila infection in aquaculture.
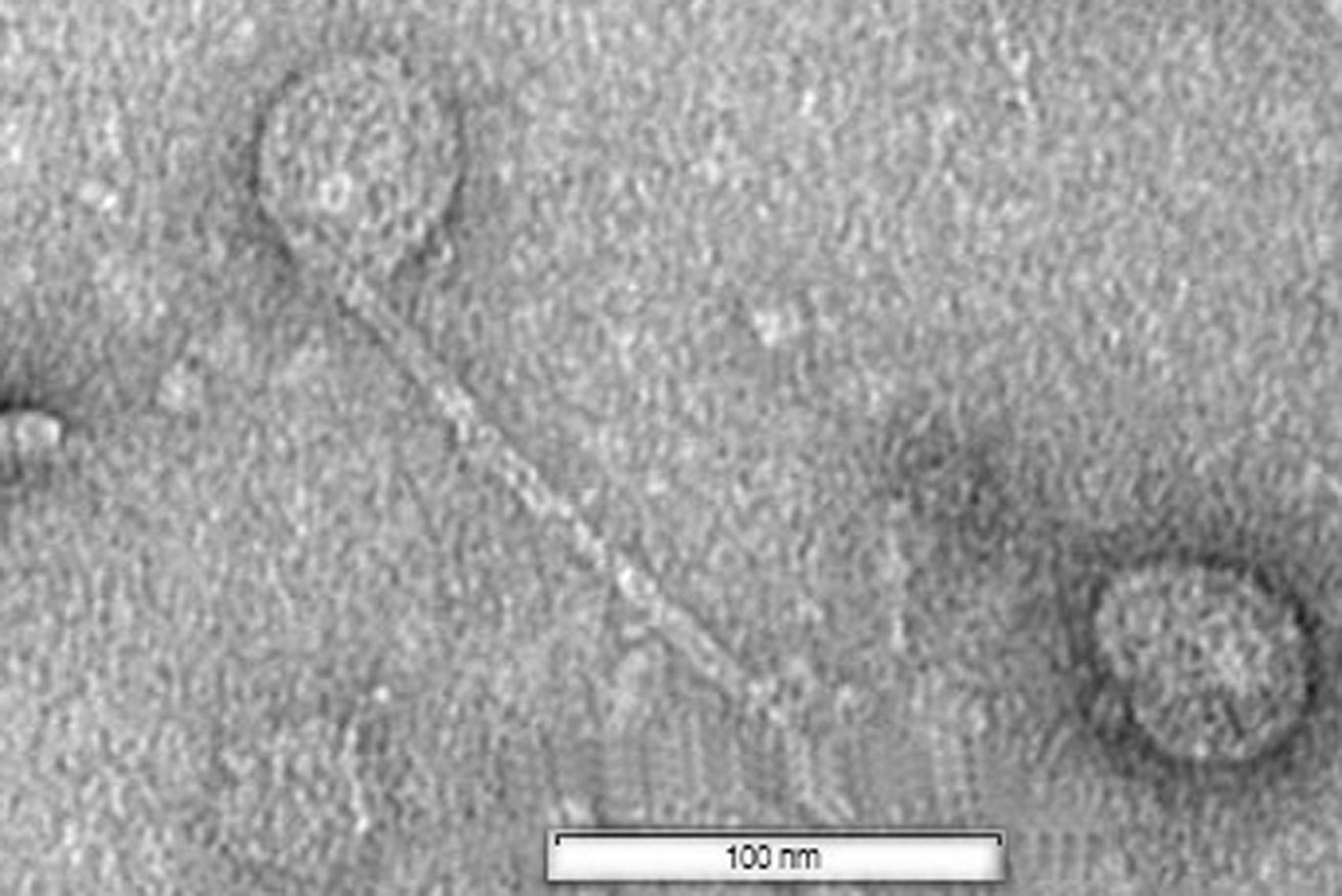
Transmission electron microscopy image of APT65 phage.
Discussion
Nowadays, a more holistic method is used in phage taxonomy, which is a significant departure from earlier morphology-based methods. 22 The APT65 phage belongs to the Heunggongvirae kingdom, Uroviricota phylum, Caudoviricetes class, and Lahexavirus genus, as confirmed by the ICTV (https://ictv.global) and whole genome sequence analysis (GenBank accession OP491958.1). There are four species in the Lahexavirus genus, including Lahexavirus LAh6, Lahexavirus LAh8, Lahexavirus LAh9, and Lahexavirus lv44572) (https://ictv.global). The ANI test revealed, however, that all of these species belong to the Lahexavirus genus and are the same species. They shared a nucleotide similarity of >97%, which is higher than the 96% cutoff value. In contrast, these four phages had a similarity of <66.8% with Lahexavirus APT65. Therefore, the other four species should be the same species, whereas Lahexavirus APT65 should be a distinct species.
Three phages known as N21, W3, and G65 were identified as being effective against virulent A. hydrophila. The phages obtained by TEM were classified as members of the Myoviridae family on the basis of their morphological characteristics. 23 Nonetheless, this family is not present on the ICVT list. To classify bacteriophages, TEM images or other morphological characteristics are insufficient; WGS is necessary.
In conclusion, morphological classification of bacteriophages is not enough. The APT65 phage isolated and identified in this study belongs to the Lahexavirus genus. Therefore, the APT65 phage should be classified as a Lahexavirus APT65 species. Since the APT65 phage does not contain virulence factors, antibiotic resistance genes, or phage lifestyle genes, it has the potential to be used in phage therapy against A. hydrophila.
Footnotes
Authors' Contributions
I.A. and M.T. designed the experiment. A.C., M.T., and M.A. performed experiments. I.A., A.C., M.T., and M.A. analyzed the data and contributed to draft writing. I.A. performed final writing—review and editing. Before submission, all authors evaluated and approved the article and agreed to accept responsibility for all parts of the study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This project was funded by the Ministry of Agriculture and Forestry of the Republic of Turkey (TAGEM/HSGYAD/B/21/A5/P4/2505).