
Abstract
Introduction:
The rpoCY75N mutation in the zinc-binding domain of the β′ subunit of Escherichia coli RNA polymerase blocks the RNA-based mechanism of transcription antitermination utilized by bacteriophage HK022.
Materials and Methods:
Mutant phages that overcome the block imposed by the rpoCY75N mutation are described. These phages, designated “orc” (
Results:
Reporter assays showed that the sequence originating from orc phages had significant promoter activity when compared with the equivalent sequence cloned from the parental phage.
Conclusions:
The newly created promoters facilitate the expression of phage genes that are essential for growth on the rpoCY75N strain by bypassing transcription terminators. The small plaque phenotype of orc phages, when grown on the mutant host, suggests that suppression of the rpoCY75N mutation is incomplete.
Introduction
The lytic growth of lambdoid bacteriophages is primarily controlled at the level of transcription by regulating termination and antitermination.1–3
In phage HK022, the transcription apparatus controlling the expression of early genes is converted into a termination-resistant form by structured RNA elements (putL and putR,
Genetic, biochemical, and cryo-EM evidence show that put RNA interacts directly with the β′ subunit zinc-binding domain of the host RNA polymerase.5,7 Single amino acid substitutions in this domain prevent HK022 growth by blocking antitermination. 7 The specificity of these host mutants adds further distinction between the λ and HK022 antitermination mechanisms. HK022 mutants capable of suppressing the effect of rpoCY75N were isolated soon after the RNAP mutations were characterized. It was reported that these phages formed small plaques on rpoCY75N host and plaques of normal size on wild-type cells. 7
Restriction analysis revealed that the mutants had lost DNA in the right operon between genes P and Q. At the time, there was very little information available about the HK022 genome. However, the corresponding region in lambda was known to contain the transcription termination sites TR2, TR3, and TR4.8,9 It was suggested that the HK022 deletion mutations allow phage growth by removing terminators, thereby permitting transcription of downstream phage genes in the mutant host. This was confirmed by removing TR2, TR3, and TR4 from an λ-HK022 hybrid phage. 7
In this study, we describe the isolation and characterization of a different class of phage mutants that overcome the effect of rpoCY75N. These mutants designated “orc” (
Materials and Methods
Bacterial strains, plasmids, and bacteriophages
Bacterial strains, plasmids, and bacteriophages are listed in Table 1.
Bacterial Strains, Plasmids, and Phage
Phage and bacterial growth
Bacteria were grown in Luria–Bertani (LB) broth at 37°C. Bacteriophages were grown and assayed as described. 13
Mutagenesis of bacteriophage O276
Bacteriophage O276, a hybrid of phage λ and HK022, 14 was mutagenized using UV light as previously described. 15 Mutants capable of growing on the nonpermissive host were recovered by plating the mutagenized lysate on the rpoCY75N host. Individual plaques were purified and high-titer lysates were prepared using standard procedures.
Genome sequencing
The genomic DNA of phage orcO367 was purified using the phenol–chloroform method. 16 Genomic DNA from phage orcO368 was purified using a Wizard DNA purification kit (Promega). The sequencing library of orcO367 was prepared at the Pittsburgh Bacteriophage Institute and processed on an Ion Torrent Personal Genome Machine; additional details are provided in King and Perez. 14 The sequencing library for orcO368 was prepared at the North Carolina State University core laboratory and processed on an Illumina platform. Reporter constructs were confirmed by dideoxy sequencing using an Applied Biosystems DNA analyzer.
Reporter constructs
Plasmid pRAK31, 4 a derivative of promoter probe vector pRS415, 17 was used to assess the activity and relative strength of suspected promoter sequences from the orc phages. It should be noted that strong promoters cloned into pRS415 do not appear to perturb plasmid copy number, presumably because the lac operon terminator protects the origin of replication. 17 pRAK31 contains an ∼300 base pair segment of HK022 sequence within its multiple cloning site. These HK022 sequences were removed by digesting the plasmid with EcoRI and BamHI and purifying the vector backbone by agarose gel electrophoresis.
To determine whether the substitutions at base pairs 35,702 and 35,703 in orcO367 created a promoter, a region encompassing the predicted promoter sequence from the orcO367 genome and the wild-type parent (O276) was amplified by PCR using primer pairs RK765 + RK767 (Table 2). Primers RK765 + RK770 were used to generate fusions that contain the TR4 terminator.
Oligonucleotides
Engineered restriction sites used for cloning are bolded.
To determine whether the substitutions at base pair 36,394 in orcO368 created a promoter, a region encompassing the predicted promoter sequence from orcO368 genome and the wild-type parent (O276) was PCR amplified using primer pairs RK812 + RK814 or RK815 + RK814. Different regions were amplified to ensure the essential regulatory sequences were cloned.
To examine the contribution of putative UP element sequences, fusions lacking these sequences were generated by annealing complementary oligonucleotides RK775 and RK776. The double-stranded product was digested with BamHI and EcoRI to generate the appropriate ends for ligation into the lacZ reporter vector.
All reporter fusions were verified by dideoxy sequencing and the details of their construction are available upon request.
β-galactosidase assays
β-galactosidase activity was measured on logarithmically growing cells of MC1000 transformed with the promoter probe plasmid constructs essentially as described in Miller, 18 except that the cells were permeabilized by treatment with chloroform and 0.005% sodium dodecyl sulfate.
Enzymes and reagents
The restriction enzymes and biochemical reagents used in this study were used according to the manufacturers' specifications.
Cloning and transformation procedures
Isolation of DNA fragments, cloning, and transformation were performed essentially as described by Sambrook and Russell. 19 Transformants containing the reporter plasmid were selected for ampicillin resistance and maintained on medium containing ampicillin (100 μg/mL).
Results
Generation of phage mutants
Bacteriophage O276 is a λ-HK022 hybrid phage that is primarily comprised of the λ genome. It was originally constructed to localize the antitermination elements in HK022 and its features have been thoroughly described. 14 The hybrid phage contains the immunity region of the HK022 genome and the antiterminator RNA sites, putR and putL. A lysate of bacteriophage O276 was mutagenized using UV light and mutants capable of forming plaques on the nonpermissive rpoCY75N host were selected.
The mutant phages were named “orc” to signify their ability to overcome the block to antitermination imposed by the rpoCY75N host mutation. Two of these phages, orcO367 and orcO368, were chosen for further characterization. The mutant phages produce normal looking plaques on wild-type host cells and small plaques on the rpoCY75N host (Fig. 1).
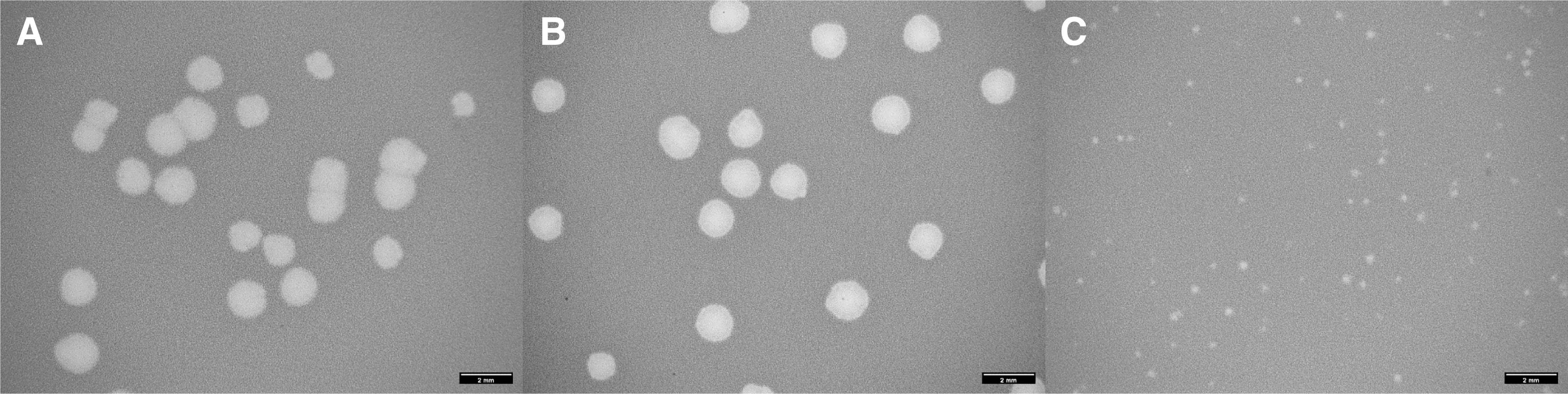
Plaque phenotype on the permissive (rpoC+) and nonpermissive (rpoCY75N) hosts.
Sequence analysis of bacteriophage orcO367
To locate the sequence changes responsible for the orc phenotype, the genomes of the mutant phages were sequenced. The genome of orcO367 differs from the parental genome at two adjacent positions (35,702 and 35,703). The base substitutions increase the similarity to the Escherichia coli σ70 promoter consensus sequence (Fig. 2).
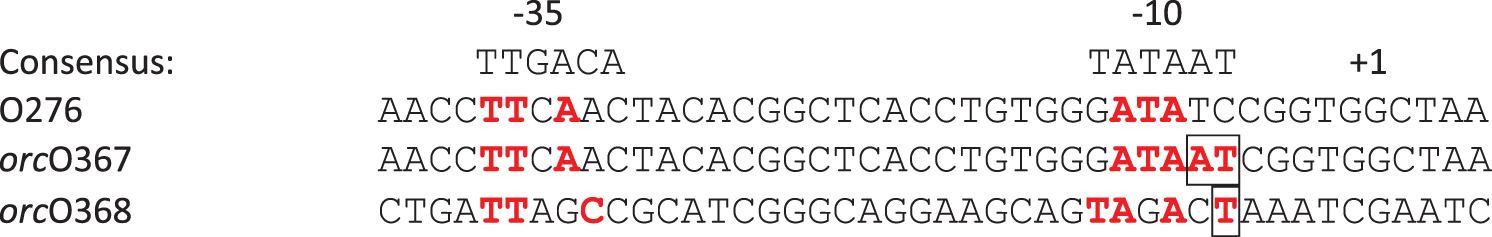
Alignment of parental (O276) and mutant (orcO367 and orcO368) phage sequences with the consensus Escherichia coli σ70 promoter hexamers. Matches to the consensus are shown in red and bolded. The orcO367 mutant has two adjacent substitutions (boxed) that improve the match to the consensus: 5 out of 6 bases in the −10 hexamer and 3 out of 6 bases in the −35 hexamer. The orcO368 mutant has a single substitution (boxed) that improves the match to the consensus: 4 out of 6 bases in the −10 hexamer and 3 out of 6 bases in the −35 hexamer. In addition, the spacing between the hexamers in the orc phage promoters differs from the consensus by a single base (18 bases vs. 17 bases [consensus]).
Sequence analysis of bacteriophage orcO368
The genome of mutant phage orcO368 differs from its parent at four positions: a single base deletion (ΔG) at position 139 (all numbering is relative to the published O276 genome), a single base addition (T) between 6130 and 6131, a single base substitution (C to T) at position 7430, and a single base substitution (G to T) at position 36,394. All four mutations were independently confirmed by amplifying the relevant regions by PCR and sequencing the products. The first two mutations were located in intergenic regions and were not predicted to impact the expression of downstream genes, and the base change at 7430 did not alter the amino acid sequence of the encoded protein.
The mutation at position 36,394 was chosen for further characterization because it also improved the match to the E. coli consensus σ70 promoter. This base change is located near a previously characterized mutation, called byp, that suppresses the effect of host nus mutants on bacteriophage λ antitermination. 20
The orc mutations create new promoters that bypass transcription terminators
The sequence analysis suggested that base substitutions identified in the orc phages created new promoter elements that drive the expression of critical phage genes. This was an attractive hypothesis since this class of mutations had been previously characterized in bacteriophage λ (see Discussion section). Similar to the λ mutants, the putative promoters in the orc phages are located downstream of well-characterized transcription terminators located in the nin region of λ genome. To test this hypothesis, we amplified the suspected sequences from the orc phages and constructed transcriptional fusions with a lacZ reporter gene. 17 For controls, similar fusions using the corresponding sequences from the wild-type parental phage (O276) were also generated.
The transcriptional fusions were electroporated into MC1000 and transformants were selected on MacConkey–lactose plates containing ampicillin. The fusions made from the wild-type phage (O276) sequences generated white colonies on MacConkey–lactose agar demonstrating a lack of promoter activity. In contrast, the fusions made from sequences amplified from orcO367 and orcO368 formed red colonies, confirming that that the base substitutions create new promoters. Quantitative β-galactosidase assays verified the indicator plate results and allowed the relative strength of the promoters to be assessed (Fig. 3).
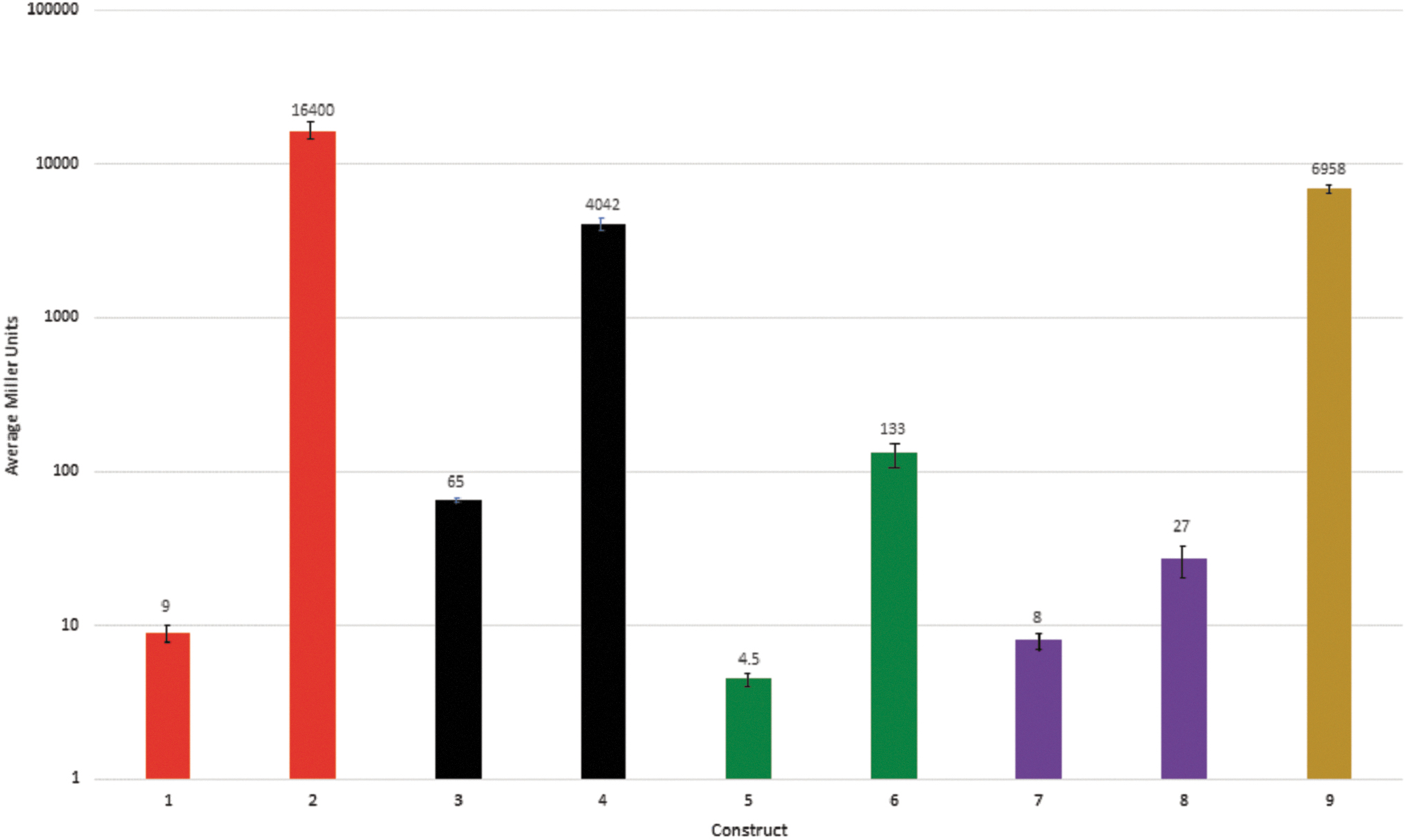
Beta-galactosidase assays of reporter constructs. Bars are colored to facilitate pairwise comparison. The numerical values for the Miller Units are shown above each bar and the error bars indicate the standard error of the mean for at least two independent measurements. Construct 1 (strain 1359) a 74 bp insert, TR4−, amplified from the wild-type phage O276, Construct 2 (strain 1360) a 74 bp insert, TR4−, amplified from the orc367 mutant, Construct 3 (strain 1362) a 564 bp insert, TR4+, amplified from the wild-type phage O276, Construct 4 (strain 1364) a 564 bp insert, TR4+, amplified from the orc367 mutant, Construct 5 (strain 1390) an 86 bp insert amplified from the wild-type phage O276, Construct 6 (strain 1387) an 86 bp insert amplified from the orcO368 mutant, Construct 7 (strain 1391) a 130 bp insert amplified from the wild-type phage O276, Construct 8 (strain 1388) a 130 bp insert amplified from the orcO368 mutant, Construct 9 (strain 1365) a 47 bp insert containing the orcO367 promoter but without the UP element, TR4−.
The steady state level of β-galactosidase activity produced from sequences originating from the parental phage (O276) was low, as expected. Conversely, the corresponding sequences from each of the orc phages produced significantly higher levels of enzyme activity. The putative promoter in orcO367 is significantly stronger than the promoter identified in orcO368 (16,400 Miller Units vs. 133 Miller Units, respectively). In addition to being more similar to the E. coli σ70 promoter consensus sequence, the promoter identified in orcO367 has an additional feature, discussed below, that contributes to its strength.
The orcO367 mutation creates a promoter that contains an UP element
Close inspection of the sequences surrounding the orcO367 promoter revealed a significant match to the E. coli UP element sequence (Fig. 4; 17 out of 22 match to the published consensus).21–22 To determine whether the UP element-like sequences contribute to promoter activity, we constructed new transcriptional fusions that lacked these sequences. E. coli cells carrying the fusion formed red colonies on MacConkey–lactose plates, indicating the deleted sequences are not essential for promoter activity. However, the quantitative β-galactosidase assays showed an approximately twofold decrease in reporter gene activity (Fig. 3: 6958 U [−UP element] vs. 16,400 U [+ UP element]). This shows that the deleted sequences do make a positive contribution to promoter activity.

Sequence similarity to the Escherichia coli UP element. The UP element consensus sequence is from Estrem et al. 22 The underlined bolded positions in the consensus can be T or A. The bases displayed in bolded red color represent the most common bases found at the indicated position. The boxed bases form the predicted −35 hexamer of the newly created promoter in orcO367.
Discussion
The control of gene expression through transcription termination and antitermination is a defining feature of bacteriophage λ (reviewed in Ref. 3 ). Upon infection, transcription initiates at the two major early promoters, PR and PL. The leftward transcript that includes the N-gene partially terminates at the TL1 terminator and approximately half the transcripts initiated at PR terminate at the TR1 terminator. The remainder of transcripts initiated at pR continue through O and P where they are terminated by terminators located in the nin region of the phage genome.
Full expression of most of early λ genes requires the binding of N protein to the phage nut sites, located within the early nascent transcripts. The N–nut complex is stabilized by several host-encoded proteins called nus factors and this complex assembly of host and phage-encoded proteins modify RNA polymerase so that pausing and termination at multiple downstream sites are suppressed. By promoting processive antitermination, transcription proceeds through multiple terminators and extends into the Q gene.1,23 The Q gene product, in turn, antiterminates transcription of the late genes.
Bacteriophage HK022, a lambdoid phage, also uses antitermination to transcribe most of its early genes.7,12 However, the modification of the elongating RNA polymerase is directly mediated by the transcripts of two closely related cis-acting phage sites, putL and putR, that modify RNA polymerase so that it reads through downstream terminators with increased efficiency.4,24–25 Like λ, the modification is stable and promotes persistent transcription antitermination over long distances.12,24,26 Unlike λ, RNA-mediated antitermination is not affected by host nus mutations. 12
Instead, antitermination is blocked by single amino acid substitutions in the β′ subunit of the host RNA polymerase. 7 The orc phages described here represent a new class of phage mutants capable of suppressing the block to RNA-mediated antitermination imposed by mutations in the β′ subunit of RNA polymerase.
The parental phage used in this study is a hybrid between λ and HK022. This phage was chosen because little is known about the identity and activity of transcription terminators in HK022. Conversely, there is a wealth of genetic and biochemical information for λ. 3 Most λ mutants exhibiting full or partial N-independence have large deletions in the pR operon (Fig. 5). 20 This class of mutations has been shown to remove multiple terminators located in the nin region of the phage. Similarly, deletions in HK022 have also been characterized and these presumably remove terminators. 7

Detailed map of the right operon of hybrid phage O276 that displays the location of mutations that suppress the requirement for transcription antitermination. The extent of deletions nin3, nin5, and roc, previously characterized in phage λ, is shown. These deletions remove terminators and allow phage growth in the absence of N and/or suppress the effect of host nus mutants. The new promoter created by the orcO367 mutation occurs just downstream of TR3 and upstream of TR4. The new promoter created by the orcO368 mutation is located downstream of TR4, similar to the λ byp mutation.
The λ byp mutation represents a different class of mutation that reduces the requirement for N-mediated antitermination. The byp mutation alone does not allow λ N− phages to grow but does allow N+λ to plate on E. coli nus host mutants. 20 The byp mutation creates a promoter that initiates transcription downstream of the terminators located in the nin region of the phage. A second mutation, c17, is required for N-independence. This mutation creates a promoter located downstream of TR1 and upstream of the replication genes O and P. 20
Similar to λ byp mutants, both orc mutants described here create new promoters. Both mutants display a small plaque phenotype even though the activity of the newly created promoters differs significantly (Fig. 3, orcO367; 16,400 U, vs. orcO368; 133 U). The small plaque phenotype suggests that orc mutations only partially suppress the effect of the rpoCY75N mutation and that the predicted increased expression of Q protein alone is insufficient to restore the wild-type plaque morphology.
We speculate that there is minimal expression of the phage replication genes in the rpoCY75N host and this reduces the contribution that gene dosage would normally provide. Although the orc mutations are in the same class as λ byp (mutations that create promoters), the orc substitutions occur at different locations and the new promoters have different activities. Only two orc mutants were characterized in this study, therefore, we cannot rule out the possibility that the base changes that occur in λ byp could also suppress the effect of rpoCY75N.
The newly identified promoter in orcO367 is separated from the Q gene by the λ TR4 terminator and our reporter assays suggest this terminator is at least 75% efficient (only 25% readthrough based on the ratio of activities measured in the presence vs. absence of the terminator [Fig. 3]; ratio of TR4+ vs. TR4−). Because transcription originating from the new promoter must traverse an efficient terminator, we suggest a strong promoter is required so that a sufficient number of transcripts reach the late gene regulator, Q.
Consistent with this hypothesis, we found the orcO367 promoter activity is strong (16,400 U) and is enhanced by the presence of UP element sequences (Figs. 3 and 4). Deletion of the UP element sequences reduced reporter activity over twofold (16,400 U vs. 6958 U). We did not attempt to engineer the UP element deletion into the parental phage, therefore, we do not know whether the UP element sequences are essential for phage growth on the rpoCY5N host. The promoter in orcO368 is weaker but there are no characterized terminators between the new promoter and Q. Therefore, transcription into Q should be unimpeded. However, it was reported that an additional weak terminator may be present between the byp mutation and Q. 20 Since our fusions do not include these sequences, we cannot evaluate any influence this putative terminator may have.
The average enzyme activity for RK1388 was significantly lower than the enzyme activity of RK1387 (27 and 133 U, respectively). The fivefold difference in enzyme activity was unexpected since both constructs contain the same promoter sequences derived from orcO368 (Fig. 3). However, these fusions contain different amounts of downstream sequence: 18 versus 62 bases from the predicted start site of transcription. We suggest that the longer RNA may fold to occlude the SD sequence and/or the start codon of the lacZ reporter. Although we have no independent evidence, this possibility is supported by predicted RNA structures generated with Mfold (data not shown). 27
We have previously reported that zinc-binding domain substitutions can distinguish between different put RNAs.6,28 Such allele specificity supports a direct and specific interaction with RNAP. Conceivably, we could have recovered put mutations that suppress the effect of the rpoCY75N. Since only two mutant phages were characterized at the sequence level, we cannot rule out that put site mutants exist in the population that formed plaques on rpoCY75N. However, this class of mutants is likely to be exceedingly rare or impossible to recover using this approach.
This is primarily due to the small size of the target (put RNAs are ∼70 bases in length) and the complexity of the put RNA structure. Put activity is strongly dependent on its structure and is less dependent on the specific identity of bases that form the structure. This is supported by extensive mutational evidence and the conservation of structural features, despite sequence variation, in put RNAs found in other phages.4,6,29 Given these restrictions, we suggest the creation of new promoters that circumvent the action of terminators likely represents the dominate class of mutations recoverable by this approach.
Footnotes
Acknowledgments
We thank John Andersland, PhD, for producing the plaque pictures. We thank the Western Kentucky University (WKU) Honors College and the WKU FUSE (Faculty-Undergraduate Student Engagement) program for their support of undergraduate research. We also thank Sofia Sileo and Jennifer King for their help constructing the figures. Bacteriophage O276 and orc phages were provided by Robert Weisberg, PhD, National Institutes of Health, USA.
Authors' Contributions
R.A.K. conceived and designed the experiments, analyzed data, and drafted the article. M.B., K.B., and C.H. performed the experiments and analyzed the data. All authors edited and agreed to be accountable for all aspects of the article and approved the final version to be published.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This project received funding from the WKU Honors College to M.B. and the WKU FUSE (Faculty-Undergraduate Student Engagement) program to M.B., K.B., and C.H. Grant numbers: M.B. #16-SP217, K.B. #13-FA103, C.H. #15-SP240.