
Abstract
Introduction
Micronuclei formation in irradiated cells that provides a general indication of both structural and numerical chromosomal aberrations is direct evidence of the mutagenic effect of UV lasers. Dose-dependent induction of micronuclei was observed in cultured human skin fibroblasts exposed to 193-nm and 248-nm laser irradiation. 2 Pyrolysis products released during the vaporization of laser-irradiated tissue also induce micronuclei formation and sister chromatid exchange. 8 It is possible that mutated cells, after several other changes, may begin unrestricted growth and form a tumor. Thus, primary DNA damage caused by laser irradiation could lead to long-term biological consequences.
During tissue ablation by laser, much of the energy is utilized by the ablation process. However, some photons may be scattered and absorbed by the surrounding viable cells. It is possible, therefore, that treatment with UV laser, although avoiding gross histopathologic changes, may result in subtle alterations in DNA in the tissues surrounding the ablated site, leading to persistent cellular defects and oncogenesis. 2 In addition, heat generation and dielectric breakdown of the tissue are frequent phenomena in laser microsurgery. As a result of heat diffusion into surrounding tissue and the shock waves created by dielectric breakdown, the volume of damaged tissue is considerably larger than might be expected from the size of the laser beam. 20
The aim of this study was to investigate the possible genotoxic effect of a new-generation UV femtosecond solid-state laser considered promising for microsurgery. Bone marrow cells from mice were used to investigate laser-irradiation genotoxicity. Single-cell gel electrophoresis, or comet, assay was used for DNA damage measurement. It is known that the comet assay is a highly sensitive and valid technique for assessing DNA damage, and, in the alkaline version, reveals the presence of single-strand breaks, double-strand breaks, and alkali-labile sites. 21 –23 The advantage of the comet assay is that the damage to genetic material is apparent without cell division and the genotoxic effect can be investigated using noncultured cells.
Materials and Methods
Animals
Female BALB/C mice aged 10–12 wk and weighing 20–25 g, were used. The animals were supplied by the Animal Facility Institute of Immunology, Vilnius University. Ethical approval was given by the Committee of Lithuanian Animal Care and Use.
Laser irradiation
Mouse bone marrow cells were irradiated by the fifth harmonic of femtosecond Yb:KGW laser Pharos (Light Conversion) producing output at 205 nm (pulse duration ca. 500 fsec, repetition rate 33 kHz). An area of 0.7 by 0.7 cm was homogeneously irradiated by scanning a collimated laser beam over it, using galvanometrically controlled scanner mirrors. Transverse cross-section of the beam was 2 mm, and the energy of the impinging pulses was about 1 μJ.
Treatment and preparation protocol
Animals were sacrificed and both femurs were dissected from each animal. Bone marrow was collected by washing the femurs with 1 mL Dulbecco phosphate-buffered saline solution (DPBS; Biochrom AG) in a plastic Eppendorf microtube. The marrow was then mixed to obtain a fine suspension. A 60-μL aliquot of the suspension was transferred onto each of nine microplates and spread over an area of 0.7 by 0.7 cm. Bone marrow cells on separate plates were exposed to different doses of 205-nm laser irradiation: 0.0175 J/cm2 (exposition span 0.5 sec), 0.035 J/cm2 (1 sec) 0.105 J/cm2 (3 sec), 0.175 J/cm2 (5 sec), 0.35 J/cm2 (10 sec), 1.05 J/cm2 (30 sec), 2.1 J/cm2 (60 sec), and 4.2 J/cm2 (120 sec). Cells on the ninth plate were irradiated by a germicidal mercury lamp (wavelength range 300–580 nm) at a dosage of 1.77 J/cm2 (exposure span 30 sec) for positive control. Immediately after irradiation, 10 μL of each sample suspension was taken to assess cell viability by Trypan blue exclusion, and 50 μL of each suspension was transferred into separate Eppendorf microtubes containing 500 μL of cold DPBS. A 60-μL aliquot of unexposed suspension (control) was placed into an additional microtube and diluted with 500 μL of cold DPBS. The suspensions in all microtubes were centrifuged at 1500 g for 5 min. The resulting cell pellets were resuspended in 500 μL of cold DPBS. The suspensions then were centrifuged again and cell pellets resuspended in 1000 μL of cold DPBS. A 40-μL aliquot of suspension from each microtube was used for the comet assay, which was performed according to the procedure described by Singh and coworkers 21 and Collins, 22 but with some modification. In brief, 40 μL of suspension from each microtube was transferred onto a microscope slide precoated with 1% standard agarose (Fermentas) in DPBS, mixed with 40 μL of 1% low melting-point agarose (Carl Roth GmbH), and a cover glass was placed on top. The slides were refrigerated at 4°C until the gel layer solidified. The cover glass was removed, 80 μL of 1% low melting-point agarose in DPBS was added, and a cover glass was placed on top. The slides were again refrigerated at 4°C until the gel layer solidified. The cover glass was removed and the slides were placed into a jar containing lysis solution (pH 10) consisting of 2.5 M NaCl (Fluka), 100 mM EDTA, 10 mM Tris, 1% Triton X-100 (Carl Roth GmbH), and 10% DMSO (Sigma) for 90 min. The slides were then transferred into alkaline electrophoresis solution (pH > 13) (1 mM EDTA and 300 mM NaOH [Riedel-de Haën]) for DNA uncoiling and expression of alkaline-labile sites, where they remained for 20 min. Electrophoresis was performed at 28 V and 230 mA (0.8 V/cm) for 40 min. The slides were then placed in neutralizing solution (pH 7.5, 0.4 M Tris) for 30 min, stained with 2% ethidium bromide (Carl Roth GmbH) for 5 min, rinsed in distilled water, and covered with cover glasses. The slides analysis was performed the next day and 100 cells on each slide were scored per treatment. The computerized image analysis system Lucia was used to determine the percentage of DNA (%DNA) in the comet tail as the most useful and validated index of DNA damage. Each experiment was performed 10 times using cells from the bone marrow of different mice. The mean and standard error of the mean (SEM) of the average of the DNA damage score of 10 gels per experimental condition were calculated.
The data were processed using the InStat V2.02 (GraphPad Software) statistical package. Statistical analysis was performed using unpaired Student's t-tests. The level of significance was p < 0.05.
Results
The cytotoxicity assessment of UV femtosecond laser irradiation as an integral part of the single cell gel assay was performed using Trypan blue exclusion. The effect of laser irradiation on the viability of bone marrow cells is presented in Fig. 1. All investigated laser irradiation doses except the lowest one (0.0175 J/cm2) caused a significant decrease in the number of viable cells (p < 0.005, p < 0.0001). Cytotoxicity of irradiation increased as its intensity rose: after irradiation at 0.035, 0.105, 0.175, and 0.35 J/cm2, the number of viable cells decreased about 11%, 18%, 32%, and 47%, respectively, as compared to the number of cells in nonirradiated control samples (92.03 ± 1.28%). The three greatest doses of irradiation (1.05, 2.1, and 4.2 J/cm2) showed a similar range of cytotoxicity by decreasing viability of cells about 70%, 73%, and 75%, respectively, as compared to control samples. Germicidal mercury lamp irradiation, used for positive control (1.77 J/cm2), did not have a noticeable effect on bone marrow cell viability.
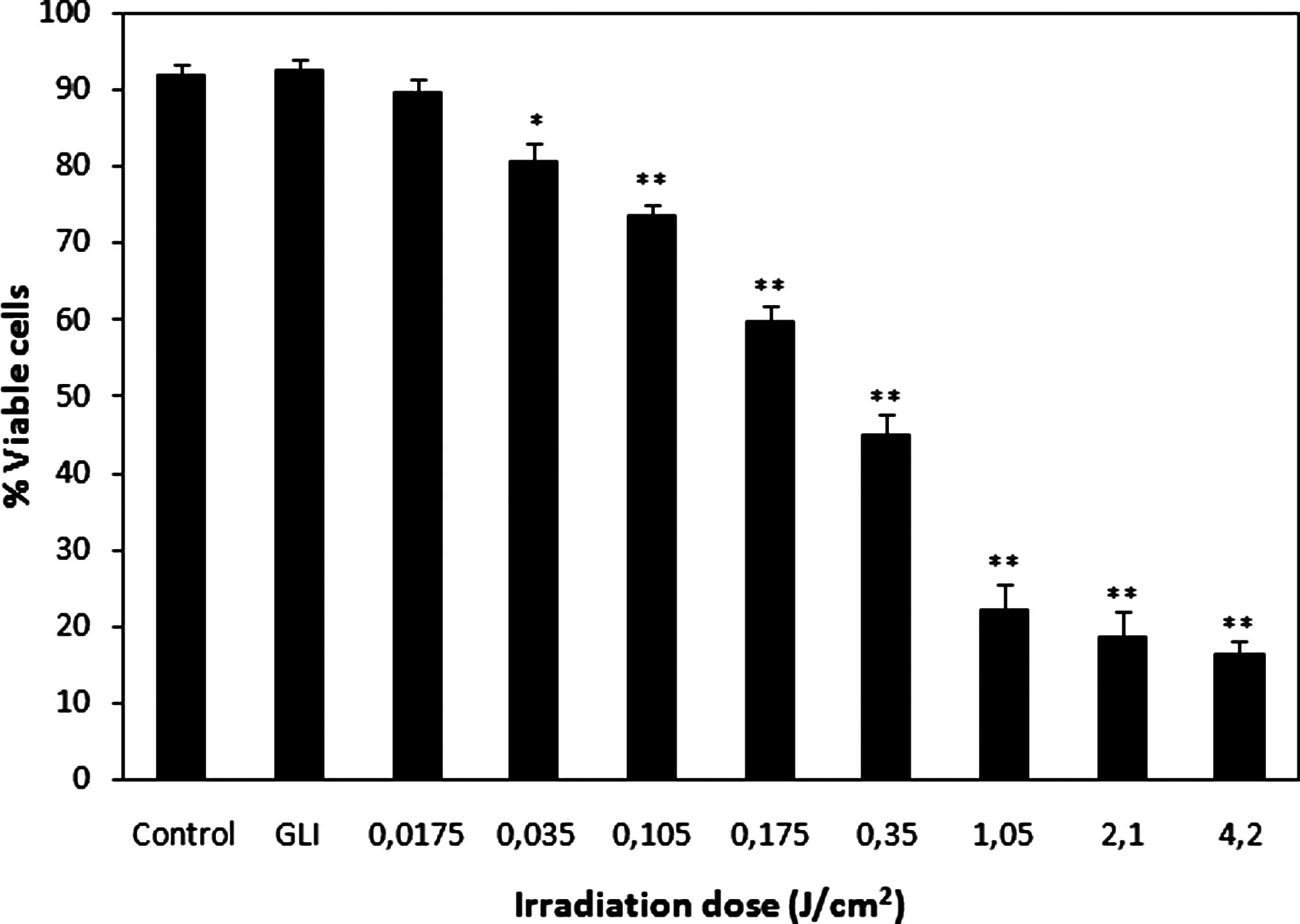
Viability of bone marrow cells exposed to 205-nm femtosecond laser irradiation at different doses. GLI denotes germicidal mercury lamp irradiation (positive control). Asterisks denote results that are significantly different from nonirradiated control (*p < 0.005; **p < 0.0001).
Figure 2 contains representative comet images of bone marrow cells exposed to different doses of 205-nm femtosecond laser irradiation and those exposed to germicidal mercury lamp irradiation (positive control), as well as an image of unexposed cells (control). The appearance of comets differed in gels on which cells were exposed to different doses of laser irradiation. Cells exposed to lower-intensity irradiation (0.0175–0.105 J/cm2) formed comets with clear large heads and comparatively small tails, whereas cells exposed to higher irradiation doses (0.175 and 0.35 J/cm2) resulted in comets with clear but smaller heads and longer, brighter tails, indicating greater damage to nuclear DNA. Most cells irradiated by the greatest doses (2.1 and 4.2 J/cm2) formed comets with small, dimmer heads and large diffuse tails. Such comets are commonly referred to as “ghost cells” (Fig. 2D) and it is known that dead or dying cells can result in comets of this type.

Representative images of murine bone marrow cell comets: after
The effect of 205-nm femtosecond laser irradiation on DNA damage in bone marrow cells assessed by comet assay is presented in Fig. 3. All investigated doses, including the lowest one, caused a significant increase in the amount of DNA in the comet tail (p < 0.0001) as compared to nonirradiated cells (3.58 ± 0.80). The percentage of tail DNA in samples irradiated at 0.0175, 0.035, and 0.105 J/cm2 (low-intensity irradiation) was 9.96 ± 0.56%, 15.13 ± 1.01%, and 26.29 ± 2.77%, respectively; in samples irradiated at 0.175 and 0.35 J/cm2 (medial-intensity irradiation), 35.96 ± 2.65% and 40.23 ± 4.21%, respectively; and in samples exposed to 1.05, 2.1, and 4.2 J/cm2 irradiation doses (high-intensity irradiation), 56.49 ± 4.69%, 60.43 ± 4.93%, and 61.06 ± 4.61%, respectively. The dosage-effect dependence was strong at the range of low-intensity irradiation (R 2 = 0.96), moderate at the range of medial-intensity irradiation (R 2 = 0.80), and weak at the range of highest irradiation doses (R 2 = 0.56). This decrease in the value of regression coefficient is consistent with the results of the cell viability assessment (Fig. 1) and analysis of comet appearance in gels (Fig. 2), indicating that the mechanism leading to the formation of strand breaks differs depending on irradiation intensity. The low-intensity irradiation showed a genotoxic effect by inducing DNA damage directly, medial-intensity irradiation demonstrated both genotoxic and cytotoxic effects, whereas high-intensity irradiation caused the death of most cells, which led to a similar number of strand breaks in gels where cells were exposed to the three greatest doses of laser irradiation (plateau effect; Fig. 3). Germicidal lamp irradiation (positive control) showed a strong genotoxic effect resulting in large numbers of strand breaks in exposed samples (55.53 ± 1.47% DNA in the comet tail).
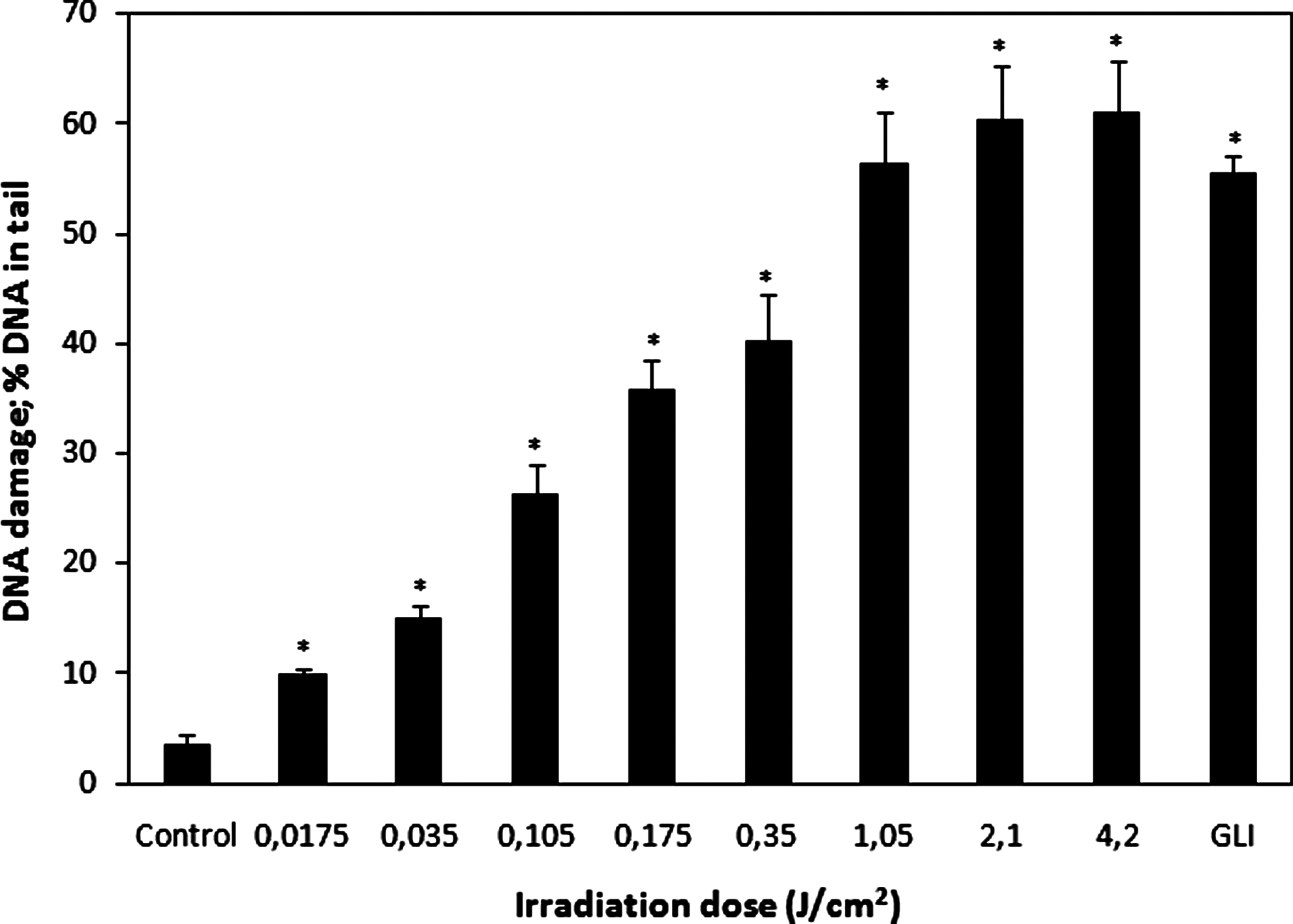
The DNA-damaging effect of 205-nm femtosecond laser irradiation, assessed by DNA content in the comet tail in bone marrow cells. GLI denotes germicidal mercury lamp irradiation (positive control). Asterisks denote results that are significantly different from nonirradiated control (*p < 0.0001).
Discussion
One type of light source that is being studied for its potential in microsurgery is the fifth harmonic of Yb-doped solid-state lasers, including the new femtosecond laser equipped with a fifth-harmonic generator. These laser systems produce an output around 200 nm. The ultra-short pulses of femtosecond lasers prevent heat accumulation in the exposed site and thus potentially produce less collateral damage of treated tissue. 24 Due to this feature and other physical parameters, such lasers could be successfully used for surgical treatment. However, the cytotoxic, genotoxic, and mutagenic impact of its irradiation has not yet been investigated. Our study of the laser's potential to induce such DNA damage as strand breaks in bone marrow cells is a first attempt to estimate the possible biological consequences of this laser treatment.
The 205-nm femtosecond laser irradiation investigated in the current study was split into relatively low-intensity irradiation (0.0175, 0.035, and 0.105 J/cm2), medial-intensity irradiation (0.175 and 0.35 J/cm2), and high-intensity irradiation (1.05, 2.1, and 4.2 J/cm2). The fringe exposure doses differed from each other by 240 times and were set at levels mimicking some characteristics of laser surgery. The greatest irradiation dose chosen (4.2 J/cm2, exposition span 2 min) is the upper limit for certain surgical treatments in clinics. The greater part of its energy goes to the cells of target tissues, but due to reflection, some of the energy may be absorbed by viable cells of surrounding tissues as diffuse light. The lowest irradiation dose used in this study (0.0175 J/cm2, exposition span 0.5 sec) was an attempt to simulate this situation.
Results of the current study demonstrated both cytotoxic and genotoxic impacts of 205-nm femtosecond laser irradiation on bone marrow cells. The cytotoxic effect, assessed by the Trypan blue exclusion, was strong at the range of high-intensity irradiation (less than 25% viable cells as compared to the control level), moderate at the range of medial-intensity irradiation (65–50% viable cells) and relatively weak at the range of low-intensity irradiation (98–80% viable cells). However, Trypan blue exclusion, routinely used in many laboratories as an integral part of the comet assay, actually does not measure viability but simply indicates whether cell membranes are intact. Cells with damaged membranes are Trypan blue positive, but may recover and survive. 25 This factor, and the possibility of identifying dead or dying cells on microscopic slides by their specific appearance, allowed us to perform comet assays on samples that showed slightly more than 30% cytotoxicity.
The genotoxic effect of laser irradiation at the range of low-intensity irradiation was surprisingly strong—the lowest exposure (0.0175 J/cm2) tripled the level of DNA damage compared with the control. The extent of DNA damage rose with increasing exposure doses at the range of low- and medial-intensity irradiation (R 2 = 0.84, p < 0.01) until a plateau was reached due to the cytotoxic effect of high-intensity irradiation. Since the ultra-short pulses of the femtosecond laser should not cause thermal damage of genetic material and the irradiation time for low- and medial-intensity irradiation (from 0.5 to 10 sec) was short compared with the DNA repair time, DNA damage at the range of irradiation of such intensities was obviously dependent on the total number of photons.
The comparison of our results with the results of studies investigating the genotoxic effect of broadband UV radiation on retinal pigment epithelial and corneal epithelial cells using comet assay 16,18 showed a similar or even higher potential of 205-nm femtosecond laser irradiation to induce DNA damage as UVB (280–315 nm) radiation, known to be extremely genotoxic and mutagenic. It is surprising because most photons at wavelengths around 200 nm and shorter are absorbed by cytoplasmic and membrane components before they reach the nuclear DNA. This is one of the reasons that 248- and 308-nm laser radiation is more frequently genotoxic than 193-nm radiation. 1 –3,26 Such high genotoxic potential of 205-nm femtosecond laser irradiation could be correlated with the pulse duration (ca. 500 fsec), which apparently is too short for the cell's relaxation after absorption of a photon; therefore, the second photon is absorbed by the cell in an excited state. Thus the cell absorbs a sort of double energy quantum, which is close to X-radiation.
In our study, we demonstrated that 205-nm femtosecond laser irradiation caused damage of different extents in bone marrow cells depending on irradiation intensity. It could be hypothesized that cells with irreversible structural damage or greatly damaged genetic material would die; therefore, primary genetic damage would not turn into a mutation when the cell's cycle came to an end. Less damaged cells could survive and DNA damage could turn into mutations through inaccurate repair. It has been demonstrated that excimer laser irradiation at 193 and 248 nm caused both micronuclei formation and a dose-dependent increase in micronuclei frequency. 2 Although the exact mechanism of micronuclei formation after UV laser irradiation remains unclear and may differ depending on the wavelength, the induction of micronuclei shows that laser irradiation caused such DNA damage as strand breaks, oxidized bases, cyclobutane pyrimidine dimmers, and 6,4-photoproducts and thus that laser irradiation has both genotoxic and mutagenic potential. Risk of mutation is greatest in cases in which laser irradiation is less cytotoxic but induces more primary damage of genetic material.
In summary, our investigations revealed the genotoxic effect of new-generation 205-nm femtosecond laser irradiation. The exposure of bone marrow cells to irradiation at relatively low doses induced strand breaks in the DNA of those cells. A dose-dependent increase in DNA strand breaks was observed. It is advisable to investigate the impact of this laser irradiation on cells of such tissues as the cornea and skin epithelium, because bone marrow cells, as cells of an internal organ, may be much more susceptible to UV-induced DNA breakage than those of external structures. Finally, investigations of the possible mutagenic effect of laser irradiation and its long-term consequences are needed for a better assessment of the risks associated with treatment procedures involving use of lasers.
Conclusion
We conclude that new-generation 205-nm femtosecond laser irradiation has a genotoxic effect by inducing strand breaks in the DNA of murine bone marrow cells in vitro. Research on the possible genotoxic effects of this laser on cells of such tissues as the cornea and skin epithelium in vivo is needed.
Footnotes
Acknowledgments
We thank senior specialist Liucija Simanskiene for assistance in performing some technical procedures, and we acknowledge with thanks the grant support provided by the Lithuanian State Science and Studies Foundation.
Author Disclosure Statement
Ph.D. Romas Danielius is a co-founder and paid employee of Light Conversion Ltd., Vilnius, Lithuania. Postgraduate Egle Gabryte is part-time employee of the same company. The remaining authors have no financial interest in the materials presented herein.