
Abstract
Introduction
Previous studies showed that visible light in the red spectrum range has a stimulatory effect on lymphocyte proliferation, synthesis of nucleic acids, and production of cytokines, apparently mediated by reactive oxygen species (ROS). 6 –9 In the blue range, the reported impact of visible light on lymphocytes and lymphoblastoid cells varied between a lethal effect, growth arrest, and stimulatory effects such as the induction of antioxidant enzymes. 10 –12 Indeed, exposure of cultured cells to visible light has been found to stimulate or inhibit cell proliferation and metabolism, depending mostly upon cell type and light wavelength, energy density, and power density. 13 The explanation given for these effects was a mechanism involving photosensitizing molecules in the cells, which absorb visible light and transfer it to nearby oxygen molecules, resulting in ROS formation. 14 –16 The objectives of this study were to elucidate the regulatory potential of blue light on inflammatory processes and to determine its effect on cultured splenocyte viability and cytokine release after activation with heat-killed Porphyromonas gingivalis, a gram-negative microorganism associated with periodontal disease. We also sought to investigate whether these effects are mediated via ROS formation.
Materials and Methods
Splenocyte isolation
Female BALB/c mice, 4–5 weeks old, were bred at The Hebrew University Specific Pathogen Free Facility. Animal use followed a protocol approved by The Hebrew University-Hadassah Medical School Institutional Animal Care and Use Committee. The spleens were removed and placed in Hank's Buffered Salt Solution (HBSS). Following hypotonic lysis of the RBC, the splenocytes were washed twice in phosphate-buffered-saline (PBS) and suspended in RPMI 1640 media (including 5% fetal calf serum, 1% glutamine, and 1% streptomycin) (Biological Industries, Beit Haemek, Israel). The cells were inoculated into black 96 well plates (NUNC, Roskilde, Denmark) (4×105 cells/ well in 200 μ l media) and incubated at 37°C in a humidified 5% CO2 incubator for 48 h. The medium was harvested for cytokine analysis and cell viability was determined using a Cell Proliferation Kit (XTT based) (Biological Industries, Beit Haemek, Israel).
Heat-killed microorganism
P. gingivalis, strain ATCC 33277 (American Type Culture Collection, Manassas, VA) was grown on blood agar plates in an anaerobic chamber with 85% N2, 5%H2, 10% CO2. After incubation at 37°C for 2–3 days, the bacteria were inoculated into peptone yeast extract and incubated for 3–4 days under the same conditions. To obtain heat-killed bacteria, the bacteria were washed 3 times with PBS and then exposed to 80°C for 10 min. 17 The bacterial concentration was standardized to an optical density of O.D.650=0.1, corresponding to ∼1010 CFU/ml. 18 The heat-killed bacteria were stored at 4°C until used, when they were re-suspended in solution by brief sonication.
Light exposure
The cells were exposed to light, using a xenon lamp with a combined filter for transmission of blue light (450–490 nm), the so-called dental plasma arc curing (PAC) light (MSq, Caesarea, Israel). An average light power of 0.345 W was measured with a power meter (Ophir, Jerusalem, Israel) over a spot of 0.7 cm diameter. The calculated power density (average power/area of the light spot) was 0.896 W/cm2.
Experimental design
Splenocytes were cultured in black 96-well plates (200 μl per well) and exposed to light with a power density of 0.9 W/cm2 for 2, 10, 30, 60, and 120 sec, equivalent to fluences of ∼2, 9, 27, 54, and 108 J/cm2, respectively. All the cells were then stimulated with 10 μl of heat-killed P. gingivalis and incubated for 48 h. Following incubation, 100 μl of media were collected from each well for analysis of cytokines IL-10, tumor necrosis factor alpha (TNFα), and interferon gamma (IFNγ). In addition, cell viability was determined using the XTT test (described subsequently). Experiments were performed in eight replicates and repeated three times. In an additional set of experiments, performed as described, ROS scavengers (as discussed subsequently) were added before exposing the cultured splenocytes to light.
Cell viability
The viability of the cells was evaluated using a colorimetric XTT assay system as described by Scudiero et al. 19 The assay is based on the ability of metabolically active cells to reduce the tetrazolium salt XTT to the orange-colored compound formazan. Briefly, after 24 h of incubation, 50 μL of XTT labeling mixture were added to each well and the microplate was incubated for a further 4 h. A Vmax microplate reader (Molecular devices, Palo Alto, CA) with a 450-nm optical filter and a 650-nm reference wavelength was used to measure the absorbance of each well. The percentage of viable cells was calculated by subtracting the optical density of the fraction of treated cells from that of the untreated cells.
Cytokine analysis
The concentration of cytokines was determined by a two-site ELISA. 20 The TNF-α and IFNγ assays were based on commercially available antibody pairs (Pharmingen, San Diego, CA). IL-10 was quantified using a commercial kit (R&D Minneapolis, MN). The 96-well ELISA plates were coated with 1 μg/ml anti-mouse cytokine monoclonal antibodies, and blocked with 3% bovine serum albumin (BSA). After adding the samples, a secondary biotinylated antibody was used as the detecting antibody, followed by a streptavidin-horseradish peroxidase conjugate (Jackson Immunoresearch Laboratories, West Grove, PA), using o-phenylenediamine (Zymed, San Francisco, CA) as substrate. The reaction was terminated by the addition of 4 N sulfuric acid, and the optical density was read with the aid of a Vmax microplate reader (Molecular Devices, Palo Alto, CA) at 490–650 nm.
Oxidant-scavenging capacity (OSC)
OSC was measured using the glucose oxidase luminescence cocktail, consisting of a mixture of luminol (10 μM), glucose oxidase (GO) (2.3 units/mL), sodium selenite (2 mM) and CoCl2·4H2O (10 μM).
21,22
In the presence of d-glucose (1 mg/mL) in HBSS, it generates a constant flux of hydrogen peroxide (H2O2) and hydroxyl radical (OH
ROS scavengers
To investigate the possible involvement of ROS in the mechanism involved in the effect of blue light on the cells, we used a mixture of different scavengers to inhibit ROS activity. Before exposing cultured cells to the light (as described previously), a “cocktail” of the following ROS scavangers was added. The cocktail contained (final concentration) 20 U/ml bovine liver catalase (Sigma, Steinheim, Germany), 100 mM dimethylthiourea (DMTU) (Sigma), 30 U/ml Escherichia coli SOD (Sigma), and 30 mM ascorbic acid (Sigma).
Temperature measurements
To investigate a possible photothermal effect on the cells during light exposure, the temperature of the cell culture medium was measured for each exposure. The measurements were repeated six times, before and immediately after exposure to light, using thermocouple electrodes (Almemo, Holzkirchen, Germany).
Statistical analysis
The data were analyzed using SigmaStat statistical software (Jandel Scientific, San Rafael, CA). One way repeated measure of analysis of variance (RM ANOVA) was used to test the significance of the differences among the treated groups. If significance was established, the intergroup differences were tested for significance using Student's t-test with the Bonferroni correction for multiple testing. The significance of the differences between two treatment groups was evaluated using the t-test. The level of significance was set at p<0.05. All the results are presented as the mean±standard error.
Results
Effect of light exposure on splenocyte viability
Splenocytes were exposed to light at different fluences. Although a low fluence (2 and 9 J/cm2) had no effect on splenocyte viability compared with that of the control in both groups with and without stimulation by P. gingivalis, higher fluences (27, 54, and 108 J/cm2) caused a decrease in cell viability (Fig. 1). In the absence of a P. gingivalis challenge, in the non-stimulated splenocytes a significant decrease in cell viability was observed in all tested fluences >27 J/cm2 (Fig. 1, black bars), whereas in the stimulated splenocytes the decrease was statistically significant only at 27 and 108 J/cm2 (Fig. 1, gray bars).
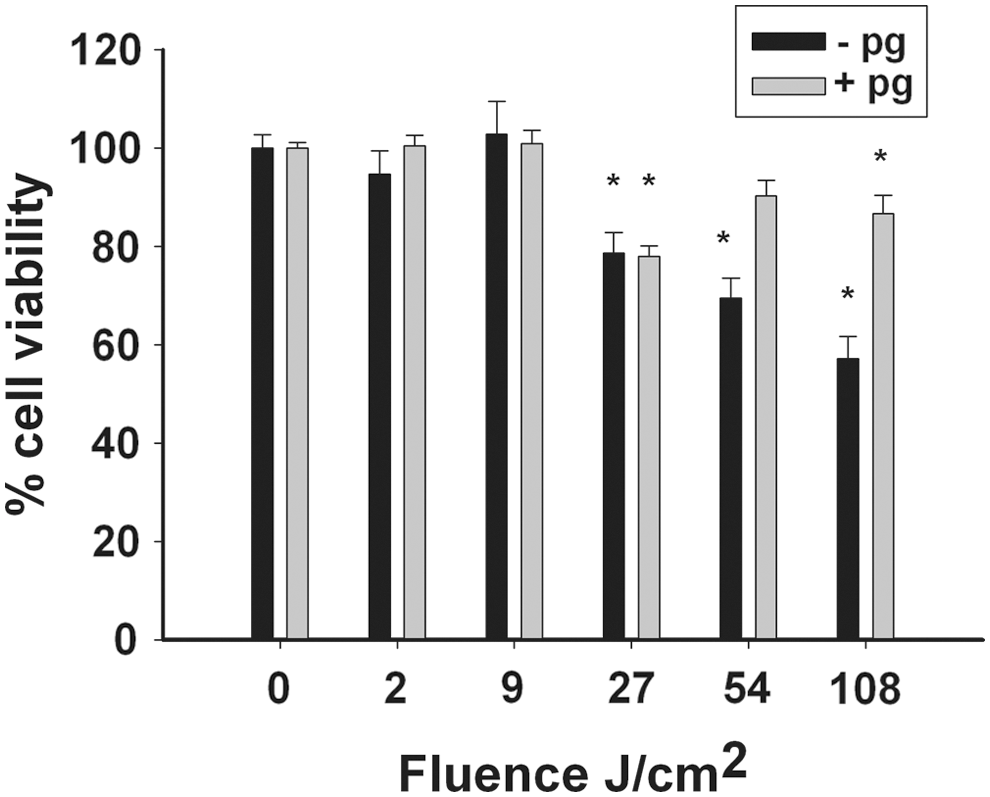
The viability of non-stimulated (black bars) and stimulated (by heat-killed P. gingivalis) splenocytes (grey bars) after exposure to blue light (0–120 sec, equivalent to fluences of 0–108 J/cm2). The viability of the cells was determined using the XTT assay. The results are expressed as mean percent cell viability (±standard error) of the non-exposed samples (100%). Significant differences between the light-exposed and the control ("0") samples are indicated by an asterisk (p<0.05, n=8 per group).
Effect of light exposure on cytokine secretion by splenocytes
Without a P. gingivalis challenge, naive splenocytes secreted a basic low level of IL-10. Following low fluence exposure (9 J/cm2), cell secretion of IL-10 increased significantly (Fig. 2a, black bars). Splenocytes stimulated by heat-killed P. gingivalis secreted a higher basic level of IL-10 compared with the baseline secretion of the naive cells (Fig. 2a, gray bars). Following low fluence exposure (2 and 9 J/cm2), cell secretion of IL-10 was significantly increased. The levels of IL-10 secretion at higher fluences (27, 54, and 108 J/cm2) were not statistically different from those of the control.
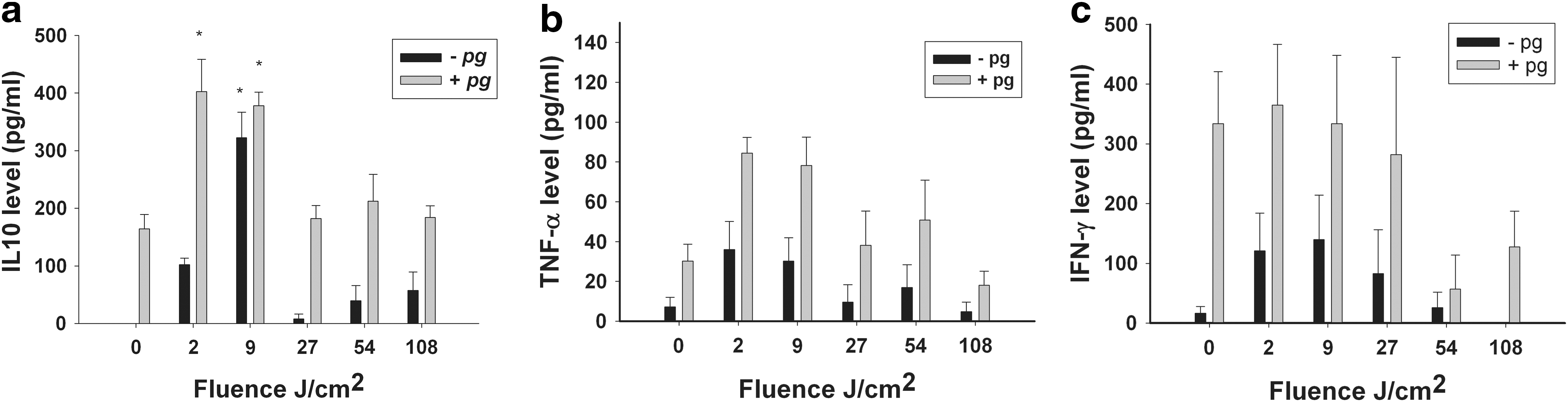
Secretion of cytokine IL-10
The levels of TNFα and IFNγ were not affected by light stimulation at any exposure time, (no statistically significant difference, Fig. 2b and c, respectively).
Effect of light exposure on splenocyte OSC
The OSC of splenocytes following exposure to blue light (0–120 sec, equivalent to fluences of 0–108 J/cm2) measured by the LDCL assay are presented in Fig. 3. The results are expressed by the ability of splenocytes to quench the luminescence generated by the GO cocktail (see Materials and Methods section). As can be seen, control splenocytes showed baseline amounts of OSC, peaking after 60 sec of 116,000±12,150 cpm. When cells were exposed to light at fluences of 2 J/cm2 (not shown) and 9 J/cm2, no significant effect was evident compared with the control (124,000±5,100 cpm). However, when longer light exposures at fluences of 27, 54 (not shown), and 108 J/cm2 were applied, the LDCL peaks were elevated to 158,000±2880, 176,000±15,700, and 169,000±25,100 cpm, respectively, a significant difference compared with the control. This elevation means that more ROS were produced following light exposure, as detected by luminescence.

OSC of the cell culture media as determined using the LDCL
Effect of light exposure on splenocyte viability and IL-10 secretion in the presence of ROS scavengers
Light exposure in the presence (Fig. 4a) or absence (Fig. 1) of ROS scavengers had a similar effect on cell viability, and the ROS scavengers tested did not show any significant protective effect against the decrease in splenocyte viability following high fluence exposure (27–108 J/cm2).
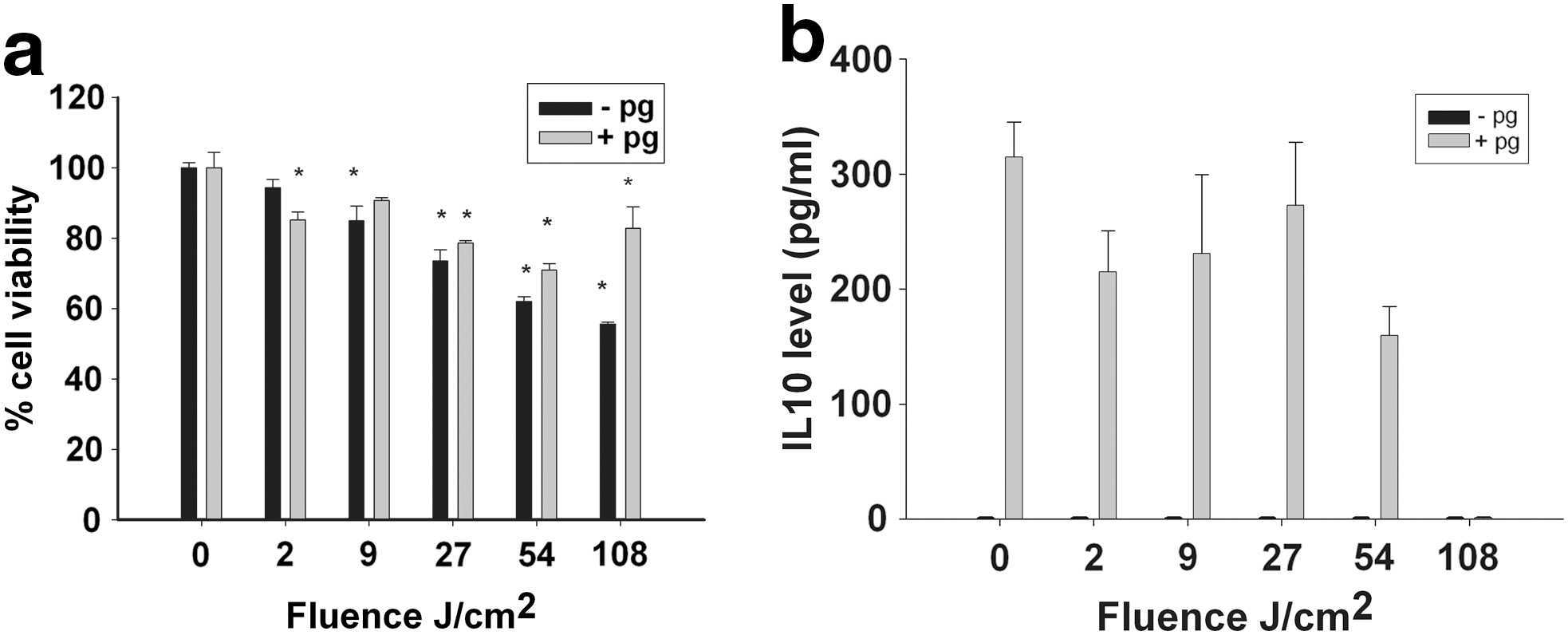
Splenocyte viability
IL-10 secretion by non-stimulated splenocytes in the presence of ROS scavengers dropped to zero, independently of light exposure (Fig. 4b, black bars). In the presence of ROS scavengers, light exposure had no significant effect on IL-10 secretion by stimulated splenocytes (Fig. 4b, gray bars). At low fluences (2 and 9 J/cm2), the levels of IL-10 were significantly lower in the presence of ROS scavengers than in their absence (Fig. 2a, gray bars).
Effect of light exposure on cell culture medium temperature
The mean temperature of the cell culture media measured immediately after light exposure was 25.5°C, 26.2°C, 28.8°C, and 30.7°C, corresponding to 2, 10, 60, and 120 sec, respectively (fluences equivalent to ∼2, 9, 54, and 108 J/cm2, respectively) (Fig. 5).
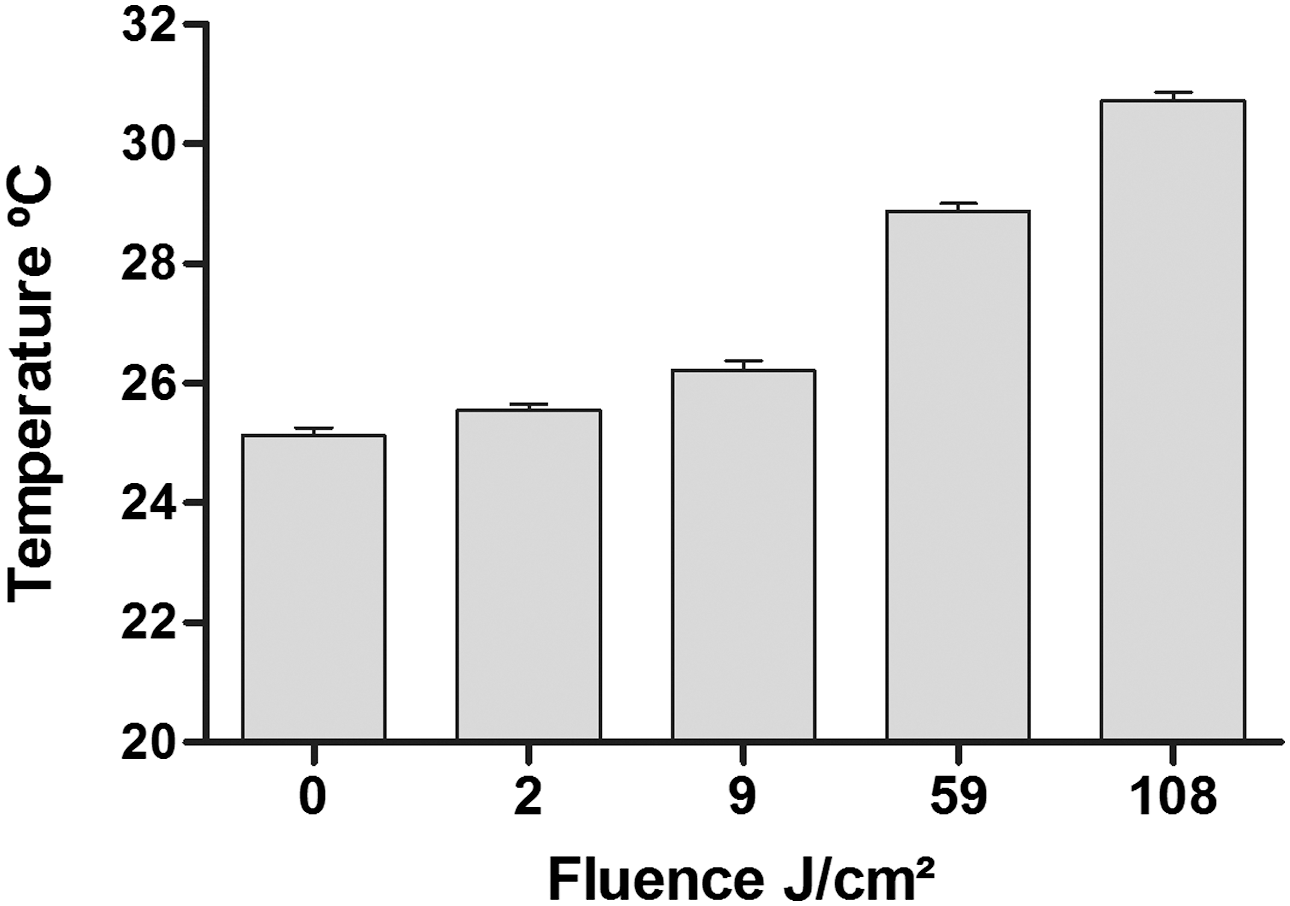
Temperature of the cell culture media measured immediately after exposure to blue light (0–120 sec, equivalent to fluences of 0–108 J/cm2). The results are expressed as the mean±standard error (n=5 per group).
Discussion
A short exposure of splenocytes to blue light at fluences of 2 and 9 J/cm2, (2 and 10 sec, respectively) had a stimulatory effect on the secretion of IL-10 (Fig. 2), which appeared to be mediated by ROS, but had no effect on the secretion of the pro-inflammatory cytokines TNFα and IFNγ or on splenocyte viability. IL-10, a cytokine with potent anti-inflammatory properties, has been implicated in the regulation of both cellular and humoral immune responses in the inflammatory process. 23 –25 It was shown that administration of IL-10 for treatment of inflammatory lesions in an animal model was short-lived and therapeutically impractical, because multiple injections were necessary to achieve modulatory effects. 26,27 Based on the present study, light may be a possible alternative noninvasive mode of therapy to enhance the IL-10 level in locally inflammatory lesions.
As is shown in Fig. 1, fluences between 54 and 108 J/cm2 (60 and 120 sec, respectively), higher than those needed to stimulate IL-10, caused a decrease in splenocyte viability with no stimulatory effect on cytokine secretion. Indeed, other studies using similar fluences also reported cytotoxic effects of blue light on other mammalian cell types. 15,28 The stimulatory effect on cells at low fluences versus cytotoxicity at higher fluences was also demonstrated by using other visible light wavelengths. Exposure of peripheral blood mononuclear cells (PBMC) to fluorescent light (380–550 nm, wavelengths) for relatively long periods (up to 4 h, equivalent to 55 J/cm2), induced the generation of antioxidant enzymes, whereas exposure for longer periods (5 h, equivalent to 68 J/cm2) resulted in enzyme inactivation probably caused by light cytotoxicity. 11 In another study, exposure of human PBMC to red light at a fluence of 18.9 J/cm2 caused an increase in the levels of all cytokines tested, whereas at a fluence of 37.8 J/cm2, the cytokine levels decreased. 7 The effect of different fluences on cell functions reported in various studies, depended on cell type, cell environment, and light parameters, such as wavelengths and fluences. 7,13,29
In the present study, we tested the effect of light on splenocytes, which are a mixture of various cell types: e.g., B cells, T cells, natural killer (NK) cells, NK T cells, dendritic cells, macrophages, and monocytes, many of which produce IL-10. 30 PBMC, used in some previous studies, are a biologically relevant system, which reflects the human inflammatory process. However, the large variation in PBMC among individuals makes this model less useful for many biological screening tests. 31 Also, there are reports that PBMC are hypersensitive to stimulation with regard to secretion of cytokines. 32 In contrast, the splenocytes derived from inbred mice (BALB/c), i.e., the mice that are genetically identical to each other, have a low variability, rendering them very useful for testing, with reproducible results. It is known that in periodontitis B cells, T cells, macrophages, and NK cells can participate in the immune response. 33 The present study shows that these cells could be affected by the light. As the immune system works as an orchestra, with cross-talk among the different cells, it was of interest to study the effect of light on the whole cell system, and then to investigate this effect on specific cell types.
Previous studies have shown that many effects of blue light on mammalian cells are mediated by ROS. 34,35 This may be explained by the presence of endogenous photosensitizers: flavins and porphyrin ring molecules concentrated in the mitochondria, which absorb visible light and transfer it to nearby oxygen molecules, resulting in ROS formation. 14 –16
ROS production appears to be light fluence-dependent. 28 Our results suggest that the enhanced secretion of IL-10 stimulated by light at low fluences might be mediated by ROS. Induction of TNFα and IFNγ, pro-inflammatory cytokines, is also known to be related to ROS. 36 The finding that no elevation of any of the tested cytokines was observed in this study could be explained by separate signal transduction pathways in different cell types. 37 In addition, IL-10 has recently emerged as an anti-inflammatory cytokine that inhibits the secretion of pro-inflammatory cytokines by monocytes and/or macrophages and the release of free oxygen radicals. 38 Acting as an antioxidant, recombinant IL-10 blocked the release of ROS in mouse peritoneal macrophages. 3 Therefore, one can postulate a balancing biofeedback between IL-10 secretion and ROS production. (a) Low levels of ROS, the production of which is stimulated by low fluences of light, enhance the secretion of IL-10, which blocks the release of ROS; and (b) the large amounts of ROS formed following exposure to high fluences, as shown by the luminescence method (Fig. 3), resulted in the depletion of IL-10 and could also contribute to the partial loss of cell viability.
However, protection against the phototoxic effect on splenocytes was not observed in the presence of ROS scavengers. The lack of protection by the scavengers could stem from their partially inefficient access to the ROS generated within the cells and their inability to scavenge the highly reactive radicals. 39 –41 Our results show that when phototoxic fluences were used, an increase in the temperature of the cell culture medium was recorded, (maximal value 30.70°C), not sufficiently high to account for the phototoxicity. However, the possibility that under certain conditions the increase in temperature could synergize with other phototoxic mechanisms, can not be ruled out.
Conclusions and Summary
Formation of the anti-inflammatory IL-10 induced by blue light may represent an adaptive cellular mechanism that might protect the cell against oxidative stress. Our study indicates the possibility of modulating the inflammatory processes by light stimulation, and suggests a potential novel treatment of inflammatory local disorders, such as periodontitis and rheumatoid arthritis. Previous studies showed that the administration of IL-10 to wild type mice significantly reduced the severity of collagen- or bacterially-induced arthritis. 42,43 IL-10 could also have a direct effect on bone homeostasis, as IL-10 may be a potent inhibitor of osteoclast formation and alveolar bone loss, 24,25,43 –46 which occur in periodontitis. Further studies, under more clinically relevant conditions, are indicated to assess the clinical significance of these promising findings.
Footnotes
Acknowledgment
This work was performed at the Ronald E Goldstein Center for Esthetic Dentistry and Dental Materials Research. The work was funded in part by the Kabacoff Foundation.
Author Disclosure Statement
No competing financial interests exist.