
Abstract
Introduction
A novel non-antibiotic approach, blue light therapy, is attracting increasing attention because of its intrinsic antimicrobial effect without the addition of exogenous photosensitizers. 10 –14 The mechanism of the antimicrobial effect of blue light is considered to be the photoexcitation of endogenous porphyrins, and, subsequently, the generation of reactive oxygen species, which are toxic to bacterial cells. 15 –17 In addition, it is commonly accepted that blue light is much less detrimental to mammalian cells than is ultraviolet irradiation, 18,19 which is another light-based antimicrobial approach being investigated. 20 Blue light has already been used in clinical applications for the treatment of inflammatory acne. 21 –24 However, the use of blue light for conventional SSTI has not been previously studied. The majority of the publications on the antimicrobial effect of blue light have been confined to in vitro studies. 10 –12,25,26 There have been (rather surprisingly) no published pre-clinical or clinical reports demonstrating blue light therapy for SSTI in animal models.
In this study, we investigated the efficacy of blue light (415±10 nm) therapy for eliminating both early-stage and established CA-MRSA infections in mouse skin abrasions. To our knowledge, this is the first in vivo study on blue light therapy for SSTI caused by CA-MRSA. We have been fortunate to be able to use bioluminescent pathogenic bacteria to develop new mouse models of SSTI. As the bacterial luminescence intensity is linearly proportional to bacterial colony-forming units (CFU), 27,28 the extent of infection can be monitored in real time by using a photon counting ICCD camera.
Materials and Methods
Bacterial strain and culture conditions
The S. aureus strain we used was USA300 LAC (Los Angeles County clone), a CA-MRSA strain. The USA300 LAC was chromosomally transduced with the transposon for the bacterial luciferase gene operon lux ABCDE (pAUL-ATn 4001 luxABCDE Km(r); Caliper Life Sciences, Hopkinton, MA) to give USA300 LAC::lux), allowing a real time monitoring of the extent of bacterial infection in living mice. 29 The bacteria were routinely grown in brain heart infusion (BHI) medium supplemented with 50 μg/mL kanamycin in an orbital incubator (37°C; 100 rpm) overnight. The overnight suspension was centrifuged, washed with phosphate-buffered saline (PBS), and re-suspended in fresh BHI medium to a defined cell density (measured by optical density) for experimental use.
Keratinocytes and culture conditions
The human keratinocyte cell line (HaCaT) 30 was cultured in 75-cm3 tissue culture flasks in 20 mL Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 μg/mL) (Sigma, St. Louis, MO). Cells were incubated at 37 °C, 95% air, 5% CO2 in a humidified incubator for 2–3 days until the cell monolayer became confluent. Growth medium was replaced every 3 days. Upon reaching at least 70% confluence, the cells were washed with PBS and trypsinized for 10 min at 37°C with 0.25% trypsin, 0.02% ethylenediamine tetraacetic acid (Sigma). The cell suspension was centrifuged, washed with PBS, and re-suspended in HEPES buffer (catalog # A14291 DJ, Life Technologies Corp., Grand Island, NY) to a defined cell density (measured by hemocytometer) for experimental use.
Light source
The light source we used was a Omnilux clear-U™ light emitting diode (LED) array (Photo Therapeutics, Inc., Carlsbad, CA) that emitted blue light at a central wavelength of 415 nm with a full width at half maximum (FWHM) of 20 nm. The irradiance of blue light on the target surface was adjusted by manipulating the distance between the LED array aperture and the target (cell culture or mouse wounds), and was measured using a PM100D power/energy meter (Thorlabs, Inc., Newton, NJ).
Blue light inactivation of MRSA in vitro
Three mL USA300 LAC::lux suspension at ≈107 CFU/mL in PBS was placed into 35 mm petri dishes at room temperature (21 °C). The suspension was irradiated with blue light LED array at an irradiance of 19.5 mW/cm2 with the lid of the petri dish removed. During the irradiation, the bacterial suspension was stirred by a mini-magnetic bar (Fisher Scientific Co., Norcross, GA). Aliquots of 40 μL of the suspension were withdrawn at 0, 24, 48, 72, 96, 120, and 144 min, respectively, when 0, 28.0, 56.1, 84.2, 112.2, 140.2, and 168.3 J/cm2 blue light had been delivered. CFU were then determined by serial dilution on BHI agar plates by the method of Jett et al. 31 Colonies were allowed to grow for 18–24 h at 37 °C. The experiments were performed in triplicate.
Blue light irradiation of keratinocytes in vitro
Three mL keratinocyte suspension at 106 cell/mL in HEPES buffer was placed into 35 mm petri dishes at room temperature (21 °C). The suspension was irradiated with the blue light LED array at an irradiance of 19.5 mW/cm2 with the lid of the petri dish removed. During the irradiation, the keratinocyte suspension was stirred by a mini-magnetic bar. Aliquots of 40 μL of the suspension were withdrawn at 0, 24, 48, 72, 96, 120, and 144 min, respectively, when 0, 28.0, 56.1, 84.2, 112.2, 140.2, and 168.3 J/cm2 blue light had been delivered. Viable counts were determined immediately by mixing each sample with an equal volume of 0.4% (w/v) trypan blue and the mixture transferred to a hemocytometer. The cell survival percentage was calculated as the ratio of the number of viable cells (unstained cells) to the total number of cells. The experiments were performed in triplicate.
Transmission electron microscopy (TEM)
Untreated and blue light treated USA300 LAC cells were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde immediately after blue light illumination, and stored overnight at 4°C. After spinning down (1200 rpm, 10 min) and decanting the fixative, 0.1 M sodium cacodylate buffer (pH 7.2) was added to the pellets. After fixation, hot agar (2% in distilled water, heated to boiling) was immediately added to each pellet. Once the agar had solidified, the cell pellets were then processed routinely, as any other tissue, for TEM. The cell pellets were postfixed in 2% OsO4 in sodium cacodylate buffer, dehydrated in a graded alcohol series, and embedded in Epon t812 (Tousimis, Rockville, MD). Ultrathin sections were cut on a Reichert-Jung Ultracut E microtome (Vienna, Austria), collected on uncoated 200 mesh copper grids, stained with uranyl acetate and lead citrate, and examined on a Philips CM-10 TEM (Eindhoven, The Netherlands). The negatives were scanned on an Epson Perfection 3200 photoscanner. Multiple parasite sections were microscopically analyzed and images representing the most typically observed morphologies were presented in the study.
Mouse model of skin abrasion infected with USA300 LAC::Lux
Animal experiments were approved by the Subcommittee on Research Animal Care (IACUC) of Massachusetts General Hospital and were in accordance with National Institutes of Health (NIH) guidelines. Adult female BALB/c mice (Charles River Laboratories, Wilmington, MA), 7–8 weeks old and weighing 16–18 g, were used. Mice were housed one per cage (to prevent attacks on wounds) and had access to food and water ad libitum. The mice were maintained on a 12 h light/dark cycle at a room temperature of 21°C.
Mice were given two intraperitoneal (i.p.) injections of cyclophosphamide (Sigma-Aldrich) before the bacterial inoculation on day 0. The first dose, 150 mg cyclophosphamide per kg mouse-body-weight (150 mg/kg) on day −4 was followed by the second dose of 100 mg/kg on day −1. This treatment reduced peripheral blood neutrophils to <100/μL-blood, fostering an environment more vulnerable to infection in the mice. 32
Before the creation of skin abrasions, mice were anesthetized by i.p. injection of a ketamine-xylazine cocktail and then shaved on the dorsal surfaces using an electric fur clipper. Mouse skin was then scraped using no. 15 scalpel blades until a reddened area appeared (just short of drawing blood). 33,34 This procedure resulted in first degree skin abrasions with most part of the epidermis removed (Fig. 1B). Each wound measured ∼1.2×1.2 cm. One drop (60 μL) of prepared bacterial suspension containing 3×106 (for mice treated at 30 min after bacterial inoculation) or 3×105 CFU (for mice treated at 24 h after bacterial inoculation) was then inoculated to the mouse wounds by using a micropipette (0–200 μL) and smeared uniformly using the micropipette tip. Higher bacterial inoculum was used for mice treated at 30 min after bacterial inoculation in order to observe a pronounce dose response of bacterial luminescence from mouse wound. The inoculum used for the mice treated at 24 h after bacterial inoculation was the minimum bacterial inoculum to develop infections in mice, as determined by preliminary experiments. In routine experiments, the bacterial densities (CFU/mL) of the bacterial suspensions were further confirmed by in vitro colony forming assay. This dual measure of viability gave additional confirmation of the reliability of results.
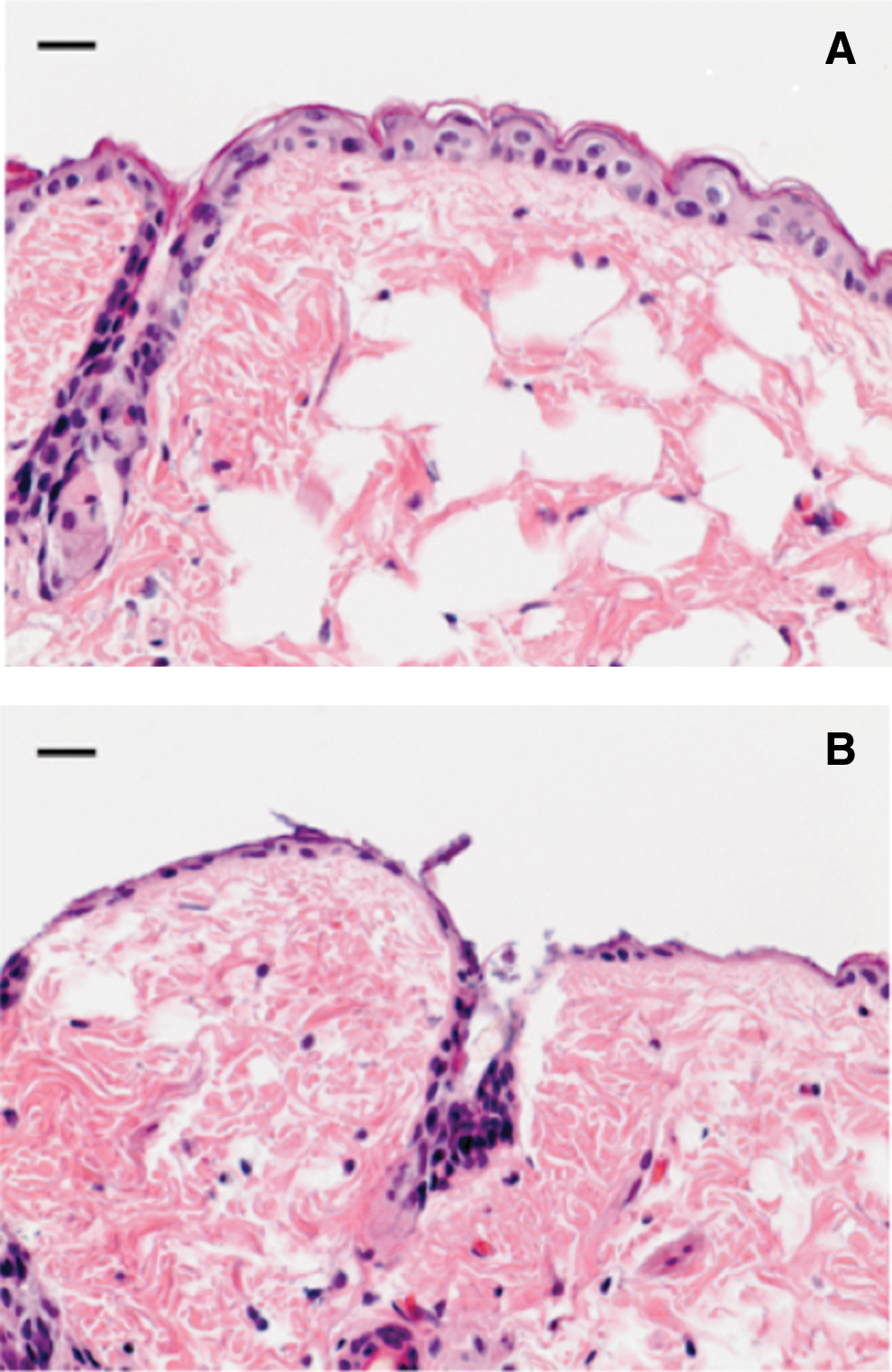
Hematoxylin and eosin (H&E) stained histology of representative BALB/c mouse skin.
Bioluminescence imaging
The setup consisted of an ICCD camera (Model C2400-30H, Hamamatsu Photonics, Bridgewater, NJ), a camera controller, an imaging box, an image processor (C5510-50, Hamamatsu), and a color monitor (PVM 1454Q, Hamamatsu). LEDs were mounted inside the imaging box and supplied the light required for obtaining dimensional imaging of the sample. Under photo counting mode, a clear image could be obtained even under extremely low light levels by detecting and integrating individual photons one by one.
Prior to imaging, mice were anesthetized by i.p. injections of ketamine/xylazine cocktail. Mice were then placed on an adjustable stage in the specimen chamber, and the infected wounds were positioned directly under the camera. A gray scale background image of each wound was made, and this was followed by a photon count of the same region. This entire wound photon count was quantified as relative luminescence units (RLU) and was displayed in a false color scale ranging from pink (most intense) to blue (least intense).
Blue light therapy for CA-MRSA infection in mouse skin abrasions
Blue light was delivered to the infected mouse wounds either at 30 min or at 24 h after the bacterial inoculation. Applying blue light at 30 min after bacterial inoculation is applicable for prophylaxis or for interrupting very early stages of infection, whereas applying blue light after a longer period will be especially important for the treatment of established infections. The irradiance used was 15.0 mW/cm2. Mice were given a total blue light exposure of up to 108 J/cm2 (120 min illumination) in aliquots, with bioluminescence imaging taking place after each aliquot of light. To record the time course of the extent of bacterial infection, bacterial luminescence from mouse wounds was recorded daily after blue light therapy until the infections were cured (characterized by the disappearance of bacterial luminescence).
Statistical analysis
The cell inactivation rates (slopes of the survival curves) and reductions in RLU from mice were compared for statistical significance using a Student t-test. p Values of<0.05 were considered significant.
Results
CA-MRSA was more susceptible to blue light inactivation in vitro than keratinocytes
The inactivation of USA300 LAC::lux in suspensions using blue light resulted in a survival curve with a shoulder or lag phase 35 (Fig. 2). Only modest inactivation (≈0.17 log10) was obtained in the lag phase until 56.1 J/cm2 blue light had been delivered. In the linear phase, the inactivation of USA300 LAC::lux by blue light approximately followed the first order kinetics, 35 with an inactivation rate coefficient of ≈0.042 cm2/J (estimated from the slope of the survival curve). Approximately 4.75-log10 bacterial inactivation was achieved in the linear phase after 112.2 J/cm2 more blue light had been delivered.
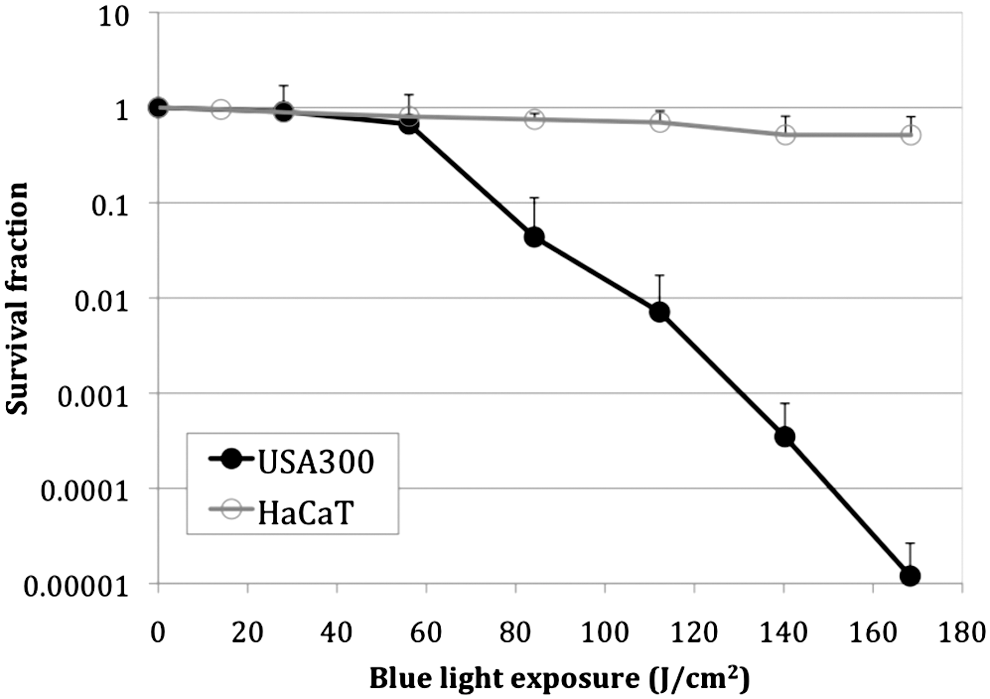
In vitro cell survival curves of USA300 LAC::lux and human keratinocyte (HaCaT) cells in response to blue light irradiation. Bars: standard deviation.
In contrast, the inactivation rate for HaCaT was much slower than that for USA300 LAC under the same blue light irradiation condition. When 168 J/cm2 blue light had been delivered, only 0.29-log10 loss of viability of HaCaT cells was observed (Fig. 2), resulting in a 4.63-log10 inactivation selectivity of USA300 LAC over HaCaT (p=0.038). The inactivation rate coefficient of HaCaT was 0.0017 cm2/J, whereas the inactivation rate coefficient of USA300 LAC in the linear phase was 0.0423 cm2/J, indicating 25-fold slower inactivation of HaCaT cells by blue light than USA300 LAC.
TEM was used to observe morphological and ultrastructural changes induced in USA300 LAC cells upon exposure to blue light (168.3 J/cm2). Disruption of the cytoplasmic contents, disruption and breakage of bacterial cell wall, and cell debris were observed in the blue light treated USA300 LAC cells (Fig. 3B). Figure 3A shows the untreated USA300 LAC cells.
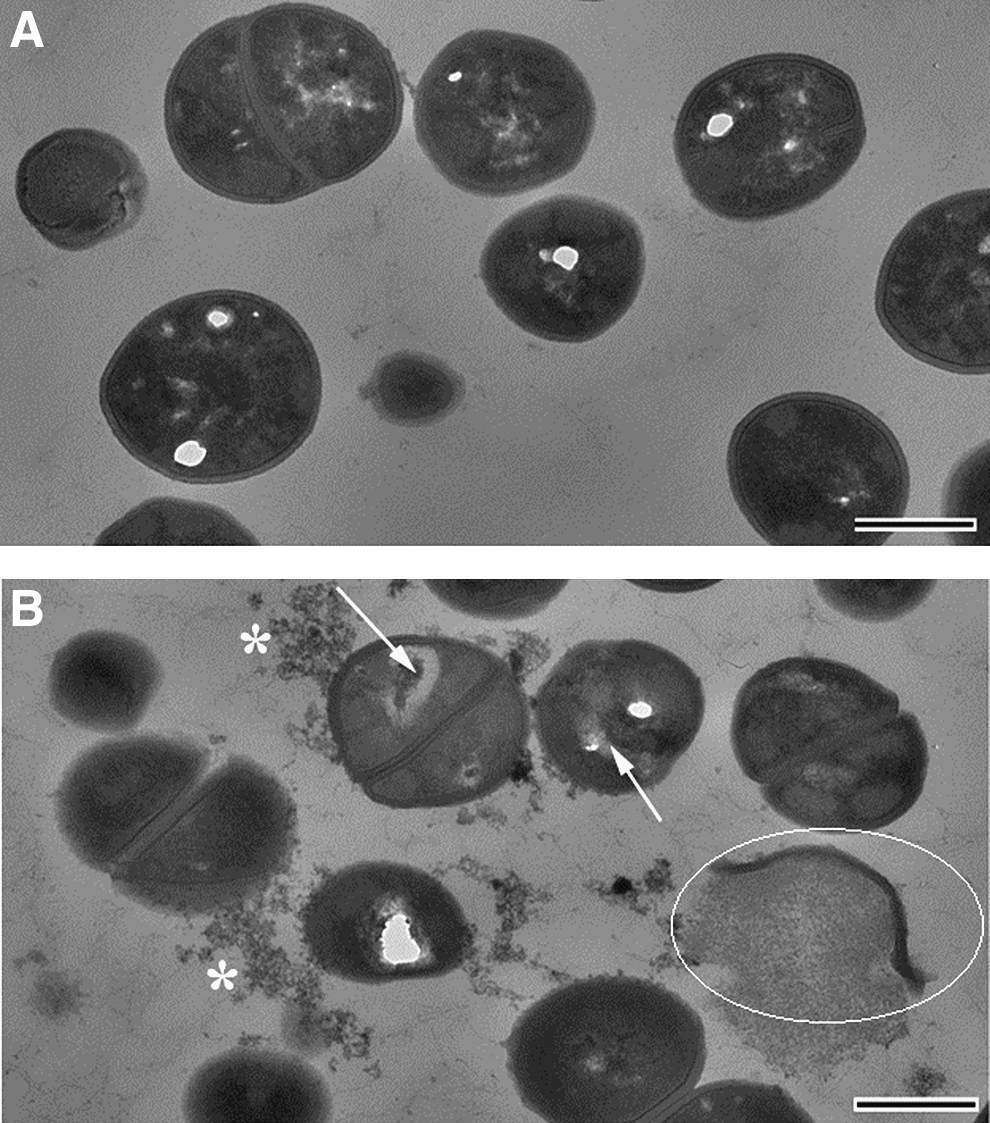
Transition electron microscopy (TEM) images of (panel
Blue light rapidly reduced the bacterial burden in both early and established CA-MRSA infections in mouse skin abrasions
Figure 4A shows the successive bioluminescence images of a representative mouse skin abrasion infected with 3×106 CFU of USA300 LAC::lux and treated with blue light (415 nm) at 30 min after bacterial inoculation (top row), and a representative mouse skin abrasion infected with 3×106 CFU of USA300 LAC::lux but without blue light treatment (bottom row). In the blue light treated wound, bacterial luminescence was almost completely eliminated after 41.4 J/cm2 blue light had been delivered (46 min illumination at the irradiance of 15.0 mW/cm2), whereas in the untreated control wound, the bacterial luminescence almost remained unchanged during the same period of time.

Blue light prophylaxis and treatment of community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) infections in mouse skin abrasions.
Figure 4B shows quantification of the RLU from the images expressed as the mean fraction of luminescence remaining for mice treated with blue light (n=4) and mice kept in the dark for the same time (n=4). The blue light values were significantly less than dark values at all fluences tested.
Figure 5A shows the successive bioluminescence images of a representative mouse skin abrasion infected with 3×105 CFU of USA300 LAC::lux and treated with blue light (415 nm) at 24 h after bacterial inoculation (top row). Bacteria multiplied ∼10-fold from 0 h (day 0) to 24 h (day 1) after bacterial inoculation as quantified by the bacterial luminescence. After 108 J/cm2 blue light had been delivered (120 min illumination at the irradiance of 15.0 mW/cm2), bacterial luminescence was almost completely eliminated. In the untreated wounds (dark control), the bacterial luminescence remained unchanged during the same period of time (bottom row). Figure 5B shows the quantification of the mean fractions of luminescence remaining as described. The fractions of luminescence remaining of the blue light group treated at day 1 (24 h post-inoculation; n=4) were significantly lower than the corresponding values of the dark control mice (n=4).

The infections on day 1 were shown to be more resistant to blue light therapy than they were on day 0. To achieve a 2-log10 inactivation of USA300 LAC:: lux in vivo, ∼ 20 J/cm2 blue light was required for day 0 treatment, whereas 95 J/cm2 was needed for day 1 treatment. The inactivation rate coefficients of bacteria in mouse skin abrasions by blue light for day 0 and day 1 treatment were ∼0.070 and 0.021 cm2/J, respectively (p=0.011).
For both blue light therapy at day 0 and day 1, bacterial regrowth, indicated by the recurrence of bacterial luminescence, was observed in mouse wounds at 24 h after blue light therapy.
Discussion
The present report is a proof of principle study on the use of blue light for CA-MRSA skin infections. Both in vitro cell culture studies and in vivo studies using mice with experimental infections were conducted.
It was found in the in vitro studies that USA 300 LAC was much more susceptible to blue light inactivation than were HaCaT cells. In the lag phase, the inactivation rate of USA300 LAC was 0.0031 cm2/J, which is only slightly higher (1.8-fold) than the inactivation rate of HaCaT (0.0017 cm2/J). However, in the linear phase, the inactivation rate of USA300 LAC dramatically increased to 0.04231 cm2/J, ∼24.8-fold higher than the inactivation rate of HaCaT cells. As a result, there existed a therapeutic window where bacteria could be selectively inactivated by blue light while the host tissue cells were able to be preserved.
The existence of the lag phase in the survival curve of USA LAC can be attributed to several causes. If clumps of bacteria existed in the suspension, all bacterial cells in the clump needed to be inactivated before the colony-forming ability of the clump was inactivated. Then a lag phase would be observed. If the lag phase represents a period in which the cells are able to resynthesize a vital component, death ensues only when the rate of destruction exceeds the rate of blue light inactivation of bacteria and is cumulative rather than instantly lethal, or there are multiple target sites for blue light inactivation, and the lag phase may also be observed. 35,36
An interesting finding from the present study is that the bacterial inactivation in vivo (in mice) using blue light was more efficient than that using bacterial suspensions in vitro. This scenario is very different from that found in many previous studies of antimicrobial photodynamic therapy (PDT), in which tens or hundreds fold higher fluences of light exposures were required for the equivalent bacterial inactivation in vivo than in vitro. 37 –39 This difference might be attributed to the in vivo environment that possibly favored the metabolism of bacterial cells compared with broth cultured cells, and promoted the biosynthesis of intracellular porphyrins, making the bacteria more photosensitive.
It was also clearly found that the infections treated on day 1 were more resistant to blue light therapy than they were on day 0. On day 1 after bacterial inoculation, the infections were fully established, and USA300 LAC existed predominantly as biofilms. The biofilm matrix could block blue light and render bacterial cells less susceptible to blue light. Additionally, when the infections were fully established, bacterial cells might have proliferated into the deeper layers of tissue such as the dermis, where the blue light transmission was attenuated.
Bacterial regrowth was observed at 24 h after blue light therapy, indicating that USA 300 LAC cells were not completely eradicated, and that the remaining amount of CFU was above the level that could induce recurrence of infection, especially in the neutropenic mice that were used in the present study. In the present study, the blue light was stopped when 2.4 and 2.2 log10 CFU reduction was achieved, respectively, for treatment at 30 min and 24 h after bacterial inoculation. In these situations, only traces of bacterial luminescence remained after blue light therapy. It was confirmed in the present study that, for the USA 300 LAC:: lux strain we used, 1.70×106 RLU luminescence corresponded to ∼3×106 CFU, that is, 0.57 RLU/CFU. At the end of blue light therapy, the remaining bacterial luminescence intensities were ∼ 1500 RLU, meaning that there were still ∼0.57×1500=900≈103 CFU remaining after blue light therapy. To prevent bacterial regrowth, especially in neutropenic mice (used to mimic immunocompromised patients) where the host defenses are absent, longer blue light illumination is required even after the bacterial luminescence is almost completely eliminated. Another option to prevent regrowth could be the possible synergistic combination of antibiotics with blue light therapy, caused by the bacteriostatic effect of antibiotics. Topical antibiotics, at a suboptimal dose that is typically found to be the case with MRSA, could be applied after blue light therapy when the majority of the bacteria were eliminated to prevent regrowth.
Footnotes
Acknowledgments
This study was supported in part by an Airlift Research Foundation Extremity Trauma Research Grant (grant #109421 to TD), a Center for Orthopaedic Trauma Advancement (COTA)/Smith & Nephew Grant (grant # 2012-16 to TD), and the NIH (grant RO1AI050875 to MRH).
Author Disclosure Statement
No conflicting financial interests exist.